

Effects of Fe and its cluster defects on laser damage of KDP and ADP crystals using first-principles
-
摘要: 由于金属杂质离子对晶体损伤性质有不容忽视的影响,受实验条件限制,Fe及其团簇缺陷对晶体的影响机制尚不明确。采用第一性原理的方法,对磷酸二氢钾(KDP)和磷酸二氢铵(ADP)晶体中的Fe及其团簇缺陷进行模拟研究,确定其对晶体结构及光学性质方面的影响。研究发现,Fe进入KDP和ADP晶体中主要以取代P原子形成FeO4基团最稳定,且其稳定形式以Fe3+为主。磁性状态研究发现磁性条件对晶体的结构和能量影响不大,Fe对晶体的损伤主要通过引起200~300 nm范围明显的光学吸收影响损伤阈值。Fe进入晶体中形成团簇缺陷可通过电荷补偿与O空位(VO)复合,几乎不会与OH空位(VOH)复合,团簇缺陷以Fe对晶体结构和性质的影响为主。Abstract: Due to the significant influence of metal impurity ions on the damage properties of crystals and the limitation of experimental conditions, the effect mechanism of Fe and its cluster defects remains unclear. In this paper, Fe and its cluster defects in KDP and ADP crystals are simulated by the method of first-principles, to determine their effects on crystal structure and optical properties. It is found that Fe atom entered into KDP and ADP crystals mainly by replacing P atom to form FeO4 group, and the relatively stable form is Fe3+. In addition, the magnetic condition has little effect on the structure and energy of the crystal, and the damage threshold of the crystal is mainly affected by the obvious optical absorption in the range of 200−300 nm. The cluster defects form when there is impurity of Fe, which could be recombined with VO through charge compensation, but hardly with VOH. The influence of the cluster defects is mainly the effect of Fe on the crystal structure and properties.
-
Key words:
- KDP crystal /
- ADP crystal /
- defect /
- laser damage /
- first-principles
-
磷酸二氢钾(KH2PO4,简称KDP)晶体具有良好的光电性能,其损伤阈值高且能够生长成大尺寸晶体,是目前唯一可用于惯性约束聚变(ICF)研究的非线性晶体材料。在工程应用中,其光学元件的实际损伤阈值远低于理论值,限制了激光的输出能量,降低了元器件的使用寿命。磷酸二氢铵(NH4H2PO4,简称ADP)晶体是KDP的同类晶体,两者具有相似的晶体结构,在四倍频方面有更广阔的应用。
缺陷是现阶段影响晶体损伤阈值的主要因素。研究表明,不仅本征点缺陷对晶体损伤有重要影响,金属杂质离子在很大程度上也会影响晶体的光学性能及损伤阈值[1-8]。2001年,Garces等研究者[9]利用电子顺磁共振的方法研究了掺Fe的KDP晶体的光学吸收和电荷转移情况,他们认为Fe在270 nm附近引入的吸收峰是由Fe3+取代K+引起,200~300 nm的吸收峰主要由Fe取代P原子形成FeO42−分子基团引起。2002年,Eremina等研究者[10-12]在晶体化学分析和KDP晶体结构模型的基础上分析了二价金属离子(Ni2+, Co2+, Fe2+, Mn2+)和三价金属离子(Al3+, Fe3+, Mn3+, Y3+, La3+)对于KDP晶体结构及光学质量的影响,他们认为二价金属离子主要影响晶体结构而三价金属离子主要影响晶体的光学吸收性质。2005年,他们利用实验方法研究了这类掺杂离子对KDP晶体生长动力学和表面形貌的影响[4]。同年,王波等研究者[13-14]研究了Fe3+对KDP晶体生长的影响,发现一定浓度的掺杂量可以提高溶液的稳定性,且Fe3+对晶体柱面的影响较大。
晶体生长的原料中存有多种微量杂质离子,Fe是最常见最具代表性的金属杂质离子,其对晶体性能的影响不容忽视。然而,由于实验技术手段的限制,Fe掺杂缺陷对晶体性能及损伤的影响机制尚不明确,实验推测并没有得到明确证实。2017年,Liu等研究者[15]利用第一性原理方法研究了掺杂Fe3+缺陷对KDP晶体的电子结构及光学性质的影响。尽管KDP晶体的此类研究取得了一定的成果,但这类点缺陷及其引入的团簇缺陷对晶体的能量、微观应力以及磁性的影响尚未可知。与此同时,ADP作为KDP晶体的同类晶体,金属离子及其团簇缺陷对其晶体结构以及性能的影响亦不容忽视,杂质离子对于两种晶体损伤阈值的影响差异也尚不清楚。因此,本文将采用第一性原理方法,探究掺杂Fe的点缺陷及其部分团簇缺陷对KDP和ADP晶体结构、能量、微观应力、电子结构和光吸收性质的影响。
1. 理论计算方法
本文采用了基于密度泛函理论的VASP软件进行理论模拟。计算中价电子和原子核的相互作用选用投影缀加波(PAW)。K、H、P、O、N、Fe原子的价电子分别为K 4s1、H 1s1、P 3s2sp3、O 2s22p4、N 2s2sp3、Fe 3d64s2,其中在点缺陷的研究中Fe原子考虑顺磁(NM)、铁磁(FM)和反铁磁(AFM)的状态[16-30]。采用Monkhorst-Pack方法选取布里渊区k空间网格点来进行积分[26]。为了提高计算效率,KDP晶体的计算参数依据文献选取,对ADP晶体的计算参数进行了收敛性测试[27-30]。对KDP晶体原有的晶格常数[31] a = b = 0.75 nm ,c = 0.697 nm和ADP晶体原有晶格常数[32] a = b = 0.75 nm,c = 0.755 nm重新定义了晶格矢量[29],即
A=(ai+aj) (1) B=(ai−aj) (2) C=ck (3) 建立含有8个KDP分子(包含64个原子)和4个ADP分子(含有48个原子)的超胞,在团簇缺陷的研究中,建立含有32个KDP分子(包含256个原子)和32个ADP分子(含有384个原子)的超胞。截断能分别设置为680 eV和400 eV。计算中电子交换相关势选用GGA-PBE泛函来优化晶体的结构[33],选用HSE06泛函来进行能量和电子性质的计算[34-35]。HSE06泛函中屏蔽距离和混合参数分别固定为1 nm和0.25。相对应点缺陷的k点[29]分别设置为4×4×4和2×2×2,团簇缺陷的k点选为1×1×1。对晶胞参数和晶胞内的原子坐标均进行了结构弛豫,达到了收敛标准使受力小于0.1 eV/nm。在ADP晶体的计算中加入范德华修正项DFT/vdW-WF2来考虑晶体中氢键的作用[36-38]。
2. 结果与讨论
室温下,KDP和ADP晶体都属于四方晶系,
D122d−I−42d 空间群[31]。为确保研究模型和结果的准确性,构建了缺陷可能存在的位点,以确定Fe原子进入KDP和ADP晶体后结构稳定的最佳位置。在模型构建中分别从晶胞中移除一个H、K和P原子,并将Fe原子添加到相应位置,除此之外,将Fe原子加入到间隙位置,分别得到Fe取代H (FeH)、Fe取代K (FeK)、Fe取代P (FeP) 和Fe间隙 (Fei) 模型,并计算缺陷形成能,如表1所示。计算缺陷形成能时,采用常温常压下稳定化合物Fe2O3来计算Fe元素的化学势,表达式为表 1 KDP晶体中Fe取代不同位点的模型体系的缺陷形成能Table 1. Defect formation energies of the KDP crystal with Fe in different locationslocation defect formation energy/eV FeH 8.7 FeP 2.74 FeK 12.59 Fei 16.10 2ΔμFe+3ΔμO=−ΔHFe2O3f (4) 式中:Fe2O3为三方晶系,空间群为R-3c。计算得到Fe的化学势为−16.40 eV。从表中可以看出,在KDP晶体中,Fe原子取代P原子形成的FeP缺陷具有最低的缺陷形成能,为2.74 eV, 明显低于Fe取代其他位置的缺陷形成能。尽管Fe与K原子的原子半径相差相对较小,但其取代K原子的缺陷形成能明显大于取代H原子的缺陷形成能。Fe原子在间隙位置形成的缺陷形成能最大,结构最不稳定,主要由于Fe原子本身的原子半径较大,在间隙位置出现引起晶体产生极大的晶格畸变致使结构不稳定。ADP是KDP的同类晶体,两者具有相似的晶体结构,通过对晶体中点缺陷形成能的计算结果可知,在ADP晶体中各缺陷的形成能大小的总体趋势与KDP晶体相同,只是具体数值有所差异,可以认为在ADP晶体中FeP缺陷也有最低的缺陷形成能,因此后续只研究了FeP缺陷。除构筑点缺陷外,在计算中考虑了Fe取代P原子形成点缺陷后与周围缺陷形成复合缺陷的情况,通过电荷补偿形成FeP缺陷与VO缺陷同时存在[32],在此基础上,通过去掉与VO缺陷相连的H原子,构成FeP与VOH缺陷同时存在的环境,计算了缺陷的形成能,如表2所示。通过结果可以看出,晶体中存在FeP与VOH团簇缺陷的几率很小,其形成能分别为24.38 eV和16.41 eV,比晶体中点缺陷的形成能高很多,因此可以认为晶体中不会存在这种形式的团簇缺陷,因此在后续计算中不予考虑。
表 2 KDP和ADP晶体中Fe团簇缺陷的缺陷形成能Table 2. Defect formation energies of the KDP crystal with Fe atom in different locationsdefect defect formation energy/eV KDP ADP VOH 8.56 0.15 FeP+VOH 24.38 16.41 FeP缺陷的取代基团结构如图1和图2所示,在KDP晶体中,移除的P原子处在PO43−骨架结构中,FeP的位置为(0.500, 0.500, 0.500),缺陷浓度为1.56%。在ADP晶体中,FeP的位置也为(0.500, 0.500, 0.500),缺陷浓度为2.08%。带电荷的缺陷模型是在中性缺陷体系中加入或去除相应数量的电子得到的,所有的结构均进行了优化。
 图 1 完美KDP和ADP晶体及其FeP缺陷的结构模型Figure 1. Crystal structures of the pristine KDP (a) and ADP (b) as well as the crystals with FeP (c, d) defects
图 1 完美KDP和ADP晶体及其FeP缺陷的结构模型Figure 1. Crystal structures of the pristine KDP (a) and ADP (b) as well as the crystals with FeP (c, d) defects 图 2 Fe替代P原子的结构示意图Figure 2. Schematic diagram of the structure for Fe replacing P atom
图 2 Fe替代P原子的结构示意图Figure 2. Schematic diagram of the structure for Fe replacing P atom从图中可以看出,KDP和ADP晶体中的化学键没有明显改变。只有处于中心区域的PO4骨架结构转变为FeO4基团结构。为分析掺杂取代之后的结构变化,研究了顺磁(NM)、铁磁(FM)和反铁磁(AFM)条件下的Fe−O键的键长变化,如表3所示。可以看出,无论是顺磁、铁磁或是反铁磁时,在同一磁性条件下随缺陷得电子数量的增加,KDP和ADP晶体中Fe−O键的键长不断增长,Fe原子与周围O原子形成的Fe−O键的键强减弱。例如,在顺磁(NM)条件下,随着FeP缺陷得电子数量增加,KDP晶体中Fe−O键的键长变化率分别为4.52%,5.16%,7.10%和9.68%。在同磁性同电荷条件下,由于晶体结构的相似性,KDP和ADP晶体中的Fe−O键的键长相差较小,仅为0.01 eV。不同磁性状态对于两晶体同电荷状态下的Fe−O键键长的影响几乎不变,可以认为磁性状态对于晶体结构几乎没有影响。可以看出在铁磁条件下KDP晶体中的FeP2−缺陷的Fe−O键键长的变化相对较大,说明在此情况下,结构产生较大的晶格畸变。
表 3 KDP和ADP晶体不同磁性条件下Fe−O键的键长变化Table 3. Fe−O bond lengths in KDP and ADP with different magnetic statesdefects bond length of Fe−O/nm KDP ADP pristine P-O 0.155 0.155 NM FeP+ 0.162 0.160 NM FeP0 0.163 0.162 NM FeP− 0.166 0.165 NM FeP2− 0.170 0.169 FM FeP+ 0.162 0.160 FM FeP0 0.163 0.162 FM FeP− 0.166 0.165 FM FeP2− 0.191 0.169 AFM FeP+ 0.162 0.161 AFM FeP0 0.163 0.162 AFM FeP− 0.166 0.165 AFM FeP2− 0.170 0.169 为进一步探究晶体中缺陷中心捕获载流子引起的能量变化,研究了不同磁性状态的带电FeP缺陷的缺陷形成能随费米能级(EF)的变化,如图3所示。图中CBM和VBM分别表示晶体的导带底和价带顶,KDP和ADP完美晶体的带隙值分别为7.00 eV和6.76 eV,费米能级在0~7.00 eV和0~6.76 eV之间变化。从图中可以看出,晶体初始状态费米能级在3.50 eV时,ADP晶体FM和AFM状态的FeP2−缺陷具有相对较低的缺陷形成能,比KDP晶体同磁性状态的FeP2−缺陷的形成能低约12 eV, 这说明FeP2−缺陷在ADP晶体中更易形成,此时Fe在晶体中为Fe3+。在KDP和ADP晶体中,不同磁性状态对应的缺陷的电荷转换能级有明显差异。例如,在KDP晶体中,NM状态下,FeP0缺陷具有最低的缺陷形成能,但其存在的范围很小,在EF = 0.10 eV时即捕获一个电子转换成FeP−缺陷,随着缺陷浓度的增加,在EF = 1.78 eV时继续捕获一个电子转换成稳定存在的FeP2−缺陷。在FM状态下,FeP−缺陷具有最低的缺陷形成能,在EF = 1.77 eV时转换成稳定存在的FeP2−缺陷。AFM状态下缺陷的转换情况与NM相似,但体系的能量低于NM状态,表现出更稳定的结构特性。在ADP晶体中,NM状态下FeP+缺陷具有相对较低的缺陷形成能,且存在范围比KDP晶体中的FeP0缺陷大,随着缺陷浓度的增加,其在EF = 2.23 eV时转换成FeP−缺陷,并在EF = 2.62 eV转换成更稳定存在的FeP2−缺陷。与KDP晶体不同,ADP晶体的FM和AFM状态的缺陷的电荷态转换情况与其NM状态相似,缺陷经+1和−1电荷态转换成稳定存在的FeP2−缺陷,FM和AFM两个状态下体系的能量相差很小,约0.14 eV。在两种晶体中,FM和AFM状态都具有最低的缺陷形成能,三种磁性状态下体系能量差异很小,差值小于1.5 eV。但磁性状态改变了KDP晶体缺陷稳定存在的状态,而对ADP晶体的缺陷电荷态没有明显影响。虽然ADP晶体更容易受到Fe掺杂缺陷的影响,但结合表3中的Fe−O键变化情况分析,Fe原子掺杂带来的ADP晶体结构的变化比KDP晶体小,尤其是掺杂取代后稳定存在的FeP2−缺陷态。因此,能量上ADP晶体内更易产生Fe原子取代缺陷,且该缺陷对晶体结构的影响小于同条件下的KDP晶体,另外,电荷转换不受磁性条件变化的影响,所以在同样出现Fe原子掺杂点缺陷时,ADP晶体表现出相对较高的晶体结构稳定性和更高抵抗损伤的能力。
 图 3 KDP和ADP晶体中不同磁性状态下FeP缺陷形成能随费米能级的变化Figure 3. Defect formation energies of the charged FeP defects in KDP and ADP as a function of Fermi energy with different magnetic states
图 3 KDP和ADP晶体中不同磁性状态下FeP缺陷形成能随费米能级的变化Figure 3. Defect formation energies of the charged FeP defects in KDP and ADP as a function of Fermi energy with different magnetic states应力是影响光学损伤的一个重要因素,而缺陷是引起微观应力的主要来源。为研究掺杂Fe缺陷带来的微观应力变化,对不同磁性条件下FeP缺陷在KDP和ADP晶体中引起的微观应力变化情况进行了研究,结果如图4所示。从图中可以看出,距离缺陷处0.5 nm左右KDP晶体中O−H键产生相对较大的局域微观应力,这种微观应力主要表现在NM状态和AFM状态中,其中AFM状态下FeP2−缺陷的O−H键变化率最大,达到50.42%,其次是AFM状态下FeP−和NM状态下FeP0缺陷,O−H键的变化率分别为48.75%和47.08%。O−H键在近邻缺陷处的变化率也相对较大,这说明Fe取代P原子破坏了PO43−骨架结构之间的连接,使相互连接的O−H键发生相对较大的形变,造成晶体结构的不稳定,通过之前能量的计算可以判断O−H键的较大形变还不至于使其发生断裂。而在ADP晶体中,O−H键的变化率小于其在KDP晶体中的值,表现出相对更强的氢键相互牵连作用,使结构稳定性受Fe原子的影响较小。但微观应力具有传播性,随着缺陷浓度的逐渐增大,微观应力不断增大、累积,会对整个晶体结构造成破坏,其影响不容忽视。与此同时,P−O键的变化率大部分不超过20%,N−H键的变化率更小,不超过10%,证明FeP缺陷对P−O和N−H键微观应力的影响较小。与本征缺陷造成的微观应力的影响相比,掺杂离子对晶体结构的影响较小,在一定程度上说明掺杂离子对晶体结构的影响不足以使晶体的抗损伤能力有较大破坏,其对晶体的影响可能体现在电子及光学性质方面。
 图 4 不同磁性状态下KDP和ADP晶体中FeP缺陷的各化学键引起的微观应力变化Figure 4. Micro-structural stress induced by various chemical bonds of FeP defect with different magnetic states in KDP and ADP crystals
图 4 不同磁性状态下KDP和ADP晶体中FeP缺陷的各化学键引起的微观应力变化Figure 4. Micro-structural stress induced by various chemical bonds of FeP defect with different magnetic states in KDP and ADP crystals为证实这一推测,计算了KDP和ADP晶体中FeP缺陷不同自旋极化状态下的电子结构及其光学性质[33-37],分态密度如图5所示,图5中插图为电荷差分密度图,黄色和蓝色分别表示电子的累积和消耗,介电函数虚部ε2(ω)如图6所示。从图5可以看出,顺磁(NM)状态下KDP晶体中FeP0缺陷在近价带顶引入孤立的未占据的杂质能级,主要由O 2p电子态和Fe 3d电子态共同作用。当缺陷捕获一个电子形成FeP−缺陷,杂质态向导带底的方向移动约1.5 eV,杂质态仍由O 2p电子态和Fe 3d电子态贡献。以上两种缺陷态均在5 eV(248 nm)和5.8 eV(213.8 nm)位置引入沿ε22方向和ε33方向的吸收峰,吸收沿晶体bc方向分布,如图6(a)所示。当缺陷继续捕获电子形成稳定的FeP2−缺陷时,杂质态移至价带顶约0.5、1.5和4 eV处,引起位于1.5 eV(826.7 nm)及2.8 eV(442.9 nm)的吸收峰,分别沿c轴和b轴方向分布。缺陷态的电子得失主要来源于近邻O与Fe之间的电子转移。在铁磁(FM)状态下,FeP−缺陷引入一个自旋向上的空杂质态,位于3 eV处,两个自旋向下的空杂质态,位于3.3和4.2 eV处。O 2p和Fe 3d电子态之间的电子跃迁引起在5 eV(248 nm)和5.8 eV(213.8 nm)位置的沿着ε22方向和ε33方向的吸收峰,吸收沿晶体的bc方向分布,如图6(a)所示,这种光吸收情况与NM状态下相同。当FeP−缺陷捕获电子形成稳定的FeP2−缺陷时,电子占据的杂质态部分移至价带顶中,其余占据的杂质态分别在2.6、3.2和4.0 eV处,杂质态同样由Fe与O原子的杂化贡献,与NM状态相同,杂质态引入1.5 eV(826.7 nm)及2.8 eV(442.9 nm)的吸收峰,分别沿c轴和b轴方向分布,但在c轴方向的吸收峰强度明显高于NM状态下c轴的吸收峰强度。在反铁磁(AFM)状态下,FeP−的缺陷态位于2.8 eV和3.8 eV,FeP−缺陷和FeP2−缺陷表现出与FM状态下相似的电子性质,FeP2−缺陷的缺陷态分别占据3.8、4.0和4.6 eV,均来自O 2p和Fe 3d电子态,O与Fe之间的杂化使晶体出现了与NM状态下类似的明显光吸收现象。以上状态中出现的较强的光学吸收将会明显影响晶体抗光损伤的能力,在强光辐照条件下极易出现损伤,严重影响晶体的性能。
 图 5 KDP和ADP晶体中不同磁性条件下FeP 缺陷电荷态的分态密度图,插图为各状态下缺陷的差分电荷密度图Figure 5. Partial density of states of KDP and ADP crystals with charged FeP defects in different magnetic states, and the insets are electronic charge differences of charged FeP defects with different magnetic states
图 5 KDP和ADP晶体中不同磁性条件下FeP 缺陷电荷态的分态密度图,插图为各状态下缺陷的差分电荷密度图Figure 5. Partial density of states of KDP and ADP crystals with charged FeP defects in different magnetic states, and the insets are electronic charge differences of charged FeP defects with different magnetic states 图 6 KDP和ADP晶体中不同磁性条件下FeP缺陷电荷态的介电函数虚部ε2 (ω)Figure 6. Imaginary part of the dielectric function ε2 (ω) of charged FeP defects with different magnetic states in KDP and ADP crystals
图 6 KDP和ADP晶体中不同磁性条件下FeP缺陷电荷态的介电函数虚部ε2 (ω)Figure 6. Imaginary part of the dielectric function ε2 (ω) of charged FeP defects with different magnetic states in KDP and ADP crystals在ADP晶体中,Fe原子掺杂缺陷表现出与KDP晶体相似的电子与光学性质的影响。在顺磁(NM)状态下,FeP+缺陷在价带顶0.5 eV及2.5 eV左右处引入由O 2p和Fe 3d电子态贡献的缺陷态,O原子与Fe原子的电子跃迁使4.5 eV(275.6 nm)及5.8 eV(213.8 nm)处出现分别沿ε22和ε33方向分布的光学吸收峰。当缺陷捕获两个电子形成FeP−缺陷时,4.3 eV处新引入一个同样由O 2p和Fe 3d电子态贡献的杂质态。从图6(b)可以看出,杂质态引起位于3.2 eV(387.5 nm)和5.5 eV(225.5 nm)沿ε22方向分布的光学吸收峰,以及位于2.5 eV(486 nm)和5.8 eV(213.8 nm)沿ε33方向分布的光学吸收峰。缺陷继续捕获电子形成更稳定的FeP2−缺陷,4.8 eV处新引入一个由O 2p和Fe 3d电子态贡献的杂质态。与KDP晶体中的NM状态类似,杂质态引入位于1.5 eV(826.7 nm)及2.8 eV(442.9 nm)的吸收峰,分别沿c轴和b轴方向分布。在铁磁(FM)状态下,FeP+缺陷引入自旋向上和自旋向下的空的缺陷态,同样来源于O 2p和Fe 3d电子态,并分别在4.5 eV(275.6 nm)和5.5 eV(225.5 nm)处引入光学吸收峰。当缺陷捕获两个电子,从图5(b)可以看出,被捕获的电子分布在缺陷中心及近邻O原子周围,缺陷态同样引入类似于FeP+缺陷的光学吸收峰。当缺陷继续捕获电子形成更稳定的FeP2−缺陷时,两对自旋平行的电子占据2.8 eV和3.2 eV的杂质态,一个自旋向下的电子占据4.1 eV的杂质态,缺陷态引入了与NM状态下相同的光学吸收峰。在反铁磁(AFM)的状态下,FeP+缺陷不断捕获电子最终形成稳定存在的FeP2−缺陷,随着缺陷捕获电子数量的增加,缺陷态的位置不断向价带顶的方向移动,Fe 3d和O 2p电子态之间的电荷转移引起的光学吸收峰不断蓝移,在晶体中形成沿bc方向分布的较大范围的光学吸收峰。
综上所述,KDP和ADP晶体中的Fe原子掺杂,在不同磁性状态下引起的缺陷态的位置略有不同,引起光学吸收峰的位置也有所差异,但缺陷态的主要来源均为O 2p和Fe 3d电子态,电子跃迁引起光学吸收的变化,致使晶体沿b和c方向200~300 nm范围出现明显的光学吸收峰,严重影响晶体的光学性能,这与文献中报道的实验值相近[9]。与KDP晶体相比,ADP晶体更易受到掺杂Fe元素的影响,表现出明显的光学吸收性质,晶体中的氢键不足以制约光学吸收在晶体中的传播,抵抗光损伤的能力下降。模拟的结果说明,即使Fe原子掺杂的ADP晶体的抗损伤能力下降,其在掺杂缺陷的情况下反映出的抵抗激光损伤的能力也高于同条件下Fe原子掺杂的KDP晶体。但Fe3+等杂质离子对晶体损伤的影响不容忽视,尤其是Fe3+杂质在晶体中与本征缺陷复合成团簇缺陷时,其对晶体结构和性质的影响也主要以Fe3+的影响为主,因此在生长和加工过程中应严格控制杂质离子的含量。
3. 结 论
本文基于第一性原理方法,研究了掺杂Fe点缺陷对KDP和ADP晶体结构和性能的影响,分析了其对晶体光损伤的影响及机理。从结构和能量的角度分析了KDP和ADP晶体中掺杂Fe点缺陷引起的结构变化和稳定性。通过理论模拟发现Fe原子进入晶体取代P原子形成FeO4基团,没有对骨架结构造成坍塌性破坏;Fe的磁性状态几乎对两晶体的结构不产生影响;与KDP晶体相比,Fe原子掺杂更易在ADP晶体中形成。 KDP晶体中距离缺陷处0.5 nm左右的O−H键具有相对较大的局域微观应力,缺陷破坏了PO43−骨架结构之间的连接,使相互连接的O−H键发生相对较大的形变,造成晶体结构的不稳定。在ADP晶体中,O−H键的变化率小于其在KDP晶体中的值,表现出相对更强的氢键的相互牵连作用,结构稳定性相对更强。尽管ADP晶体中存在大量的氢键可以牵制晶体中缺陷带来的结构变化,但掺杂缺陷对光学性质的影响却依旧存在。 KDP和ADP晶体不同磁性状态的电子性质及光吸收性质表现形式不同,但两者均引入由Fe 3d和O 2p电子态共同作用的缺陷能级,电子的跃迁引起了200~300 nm范围明显的光学吸收峰,致使晶体的光学吸收性能明显降低,使其抵抗损伤的能力下降,从而影响晶体的损伤阈值。在晶体的实际应用中,应控制晶体中杂质缺陷的含量,尽量降低杂质离子对晶体损伤阈值的影响。
-
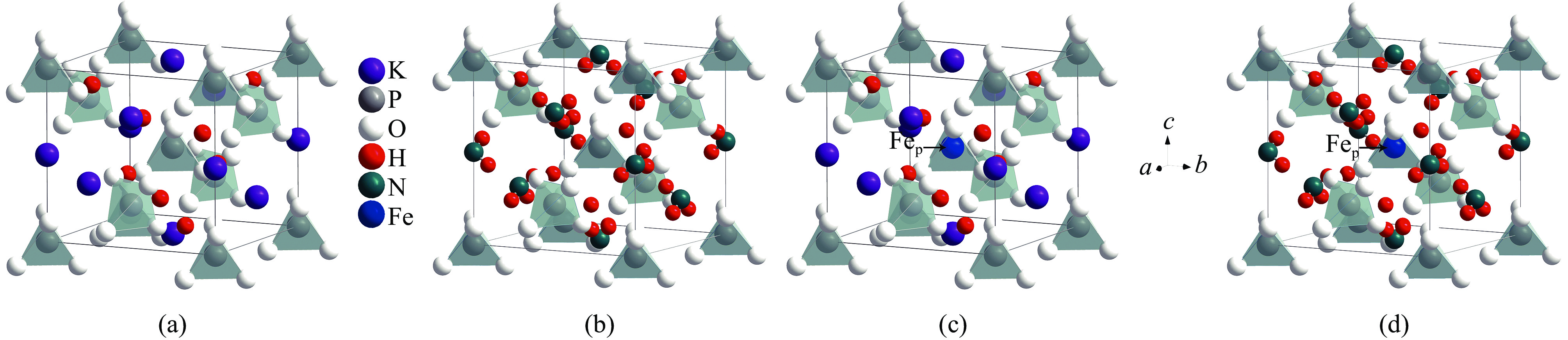
图 1 完美KDP和ADP晶体及其FeP缺陷的结构模型
Figure 1. Crystal structures of the pristine KDP (a) and ADP (b) as well as the crystals with FeP (c, d) defects
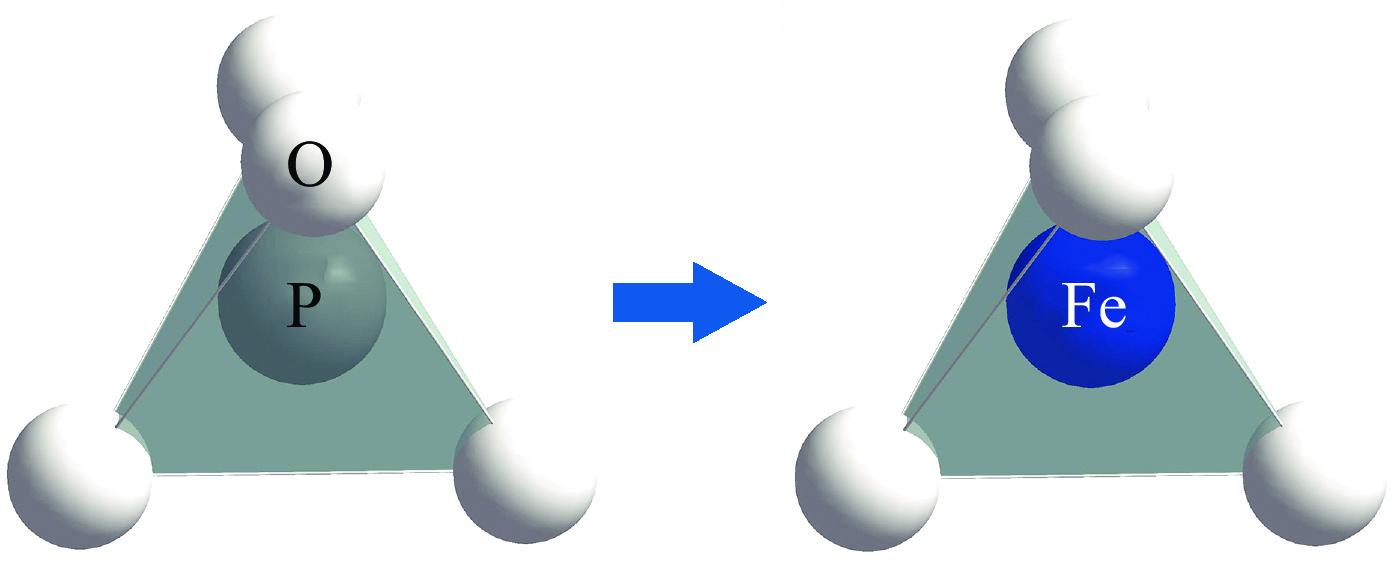
图 2 Fe替代P原子的结构示意图
Figure 2. Schematic diagram of the structure for Fe replacing P atom
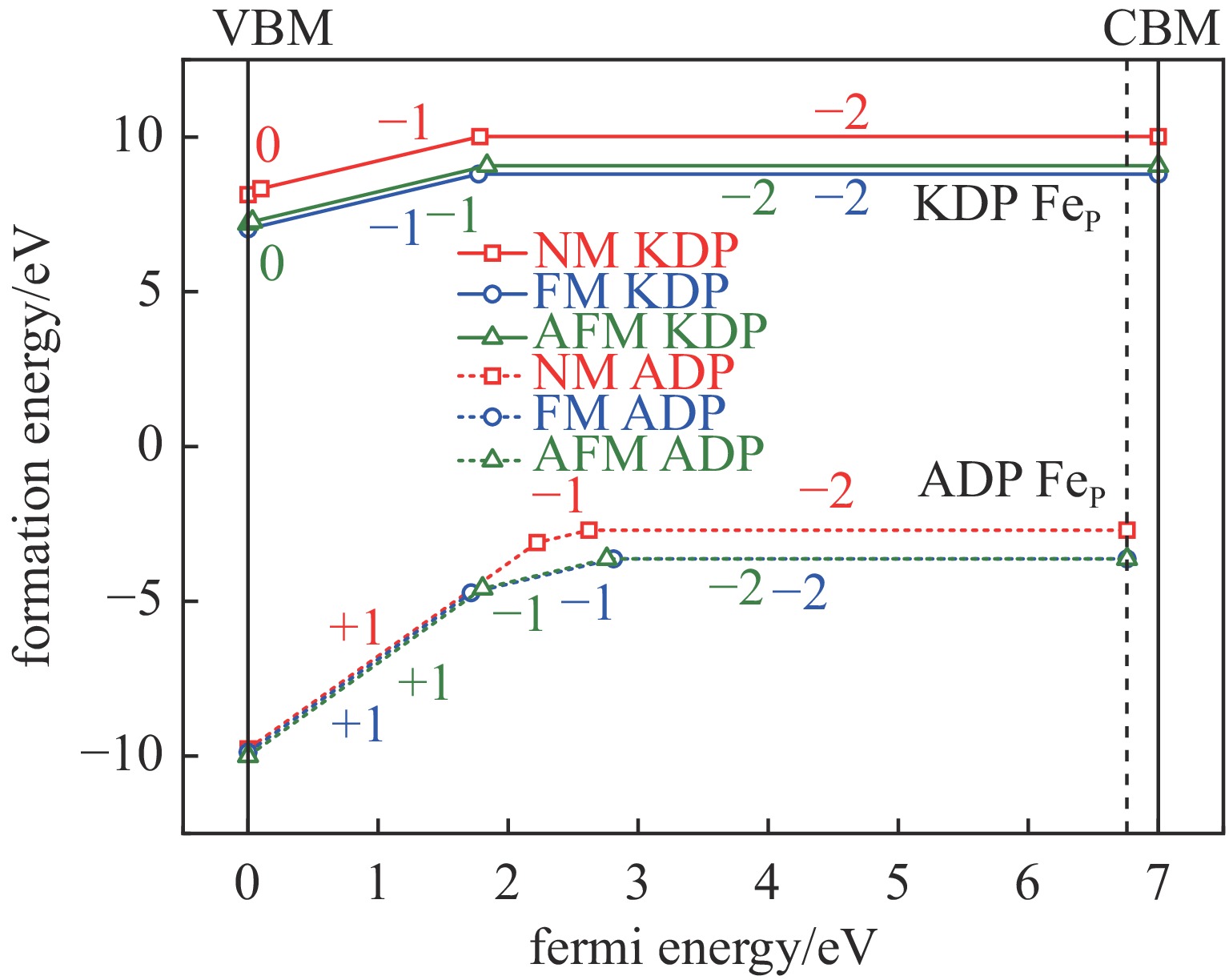
图 3 KDP和ADP晶体中不同磁性状态下FeP缺陷形成能随费米能级的变化
Figure 3. Defect formation energies of the charged FeP defects in KDP and ADP as a function of Fermi energy with different magnetic states
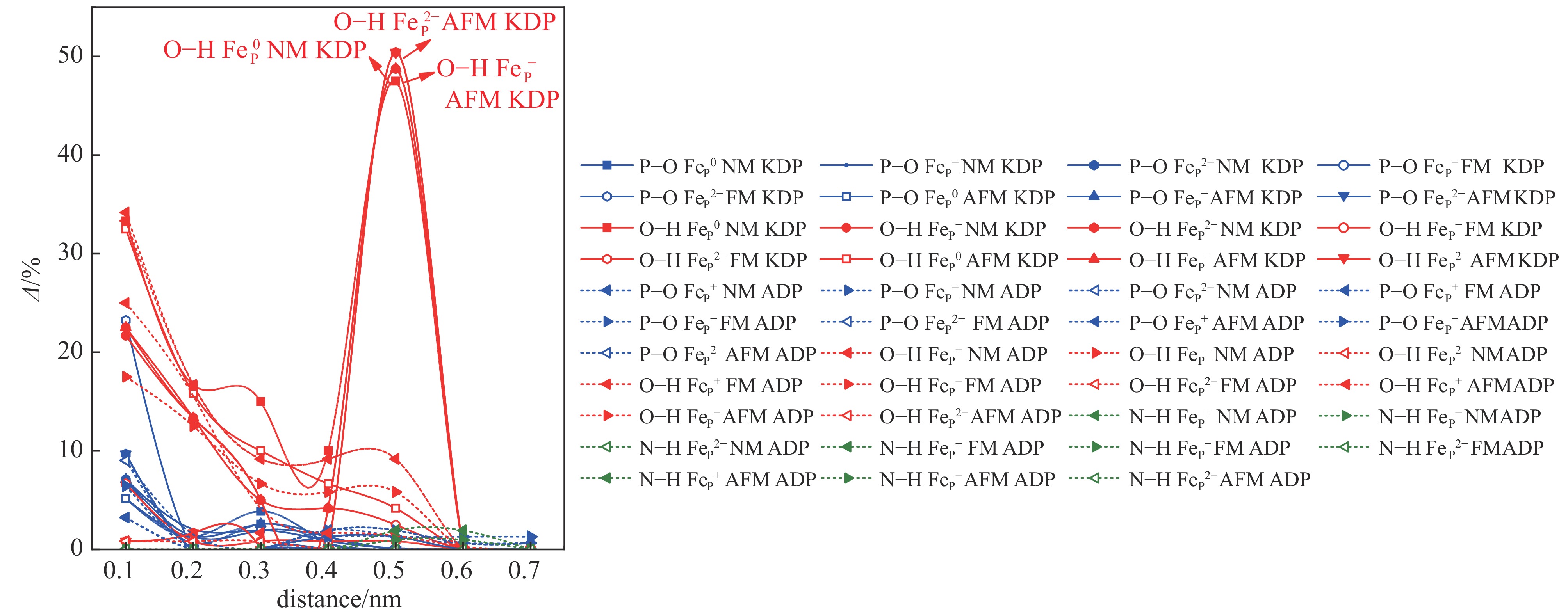
图 4 不同磁性状态下KDP和ADP晶体中FeP缺陷的各化学键引起的微观应力变化
Figure 4. Micro-structural stress induced by various chemical bonds of FeP defect with different magnetic states in KDP and ADP crystals
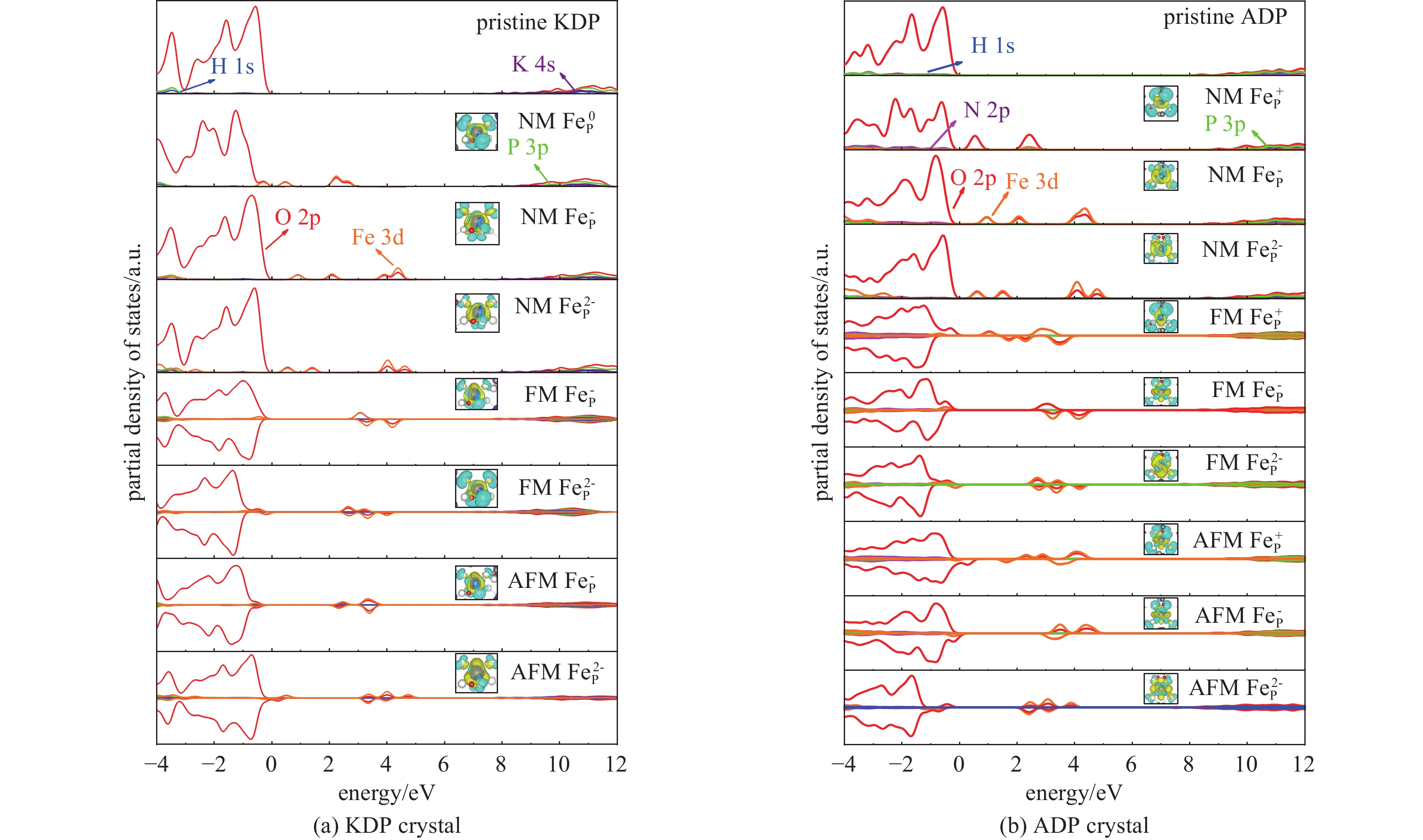
图 5 KDP和ADP晶体中不同磁性条件下FeP 缺陷电荷态的分态密度图,插图为各状态下缺陷的差分电荷密度图
Figure 5. Partial density of states of KDP and ADP crystals with charged FeP defects in different magnetic states, and the insets are electronic charge differences of charged FeP defects with different magnetic states
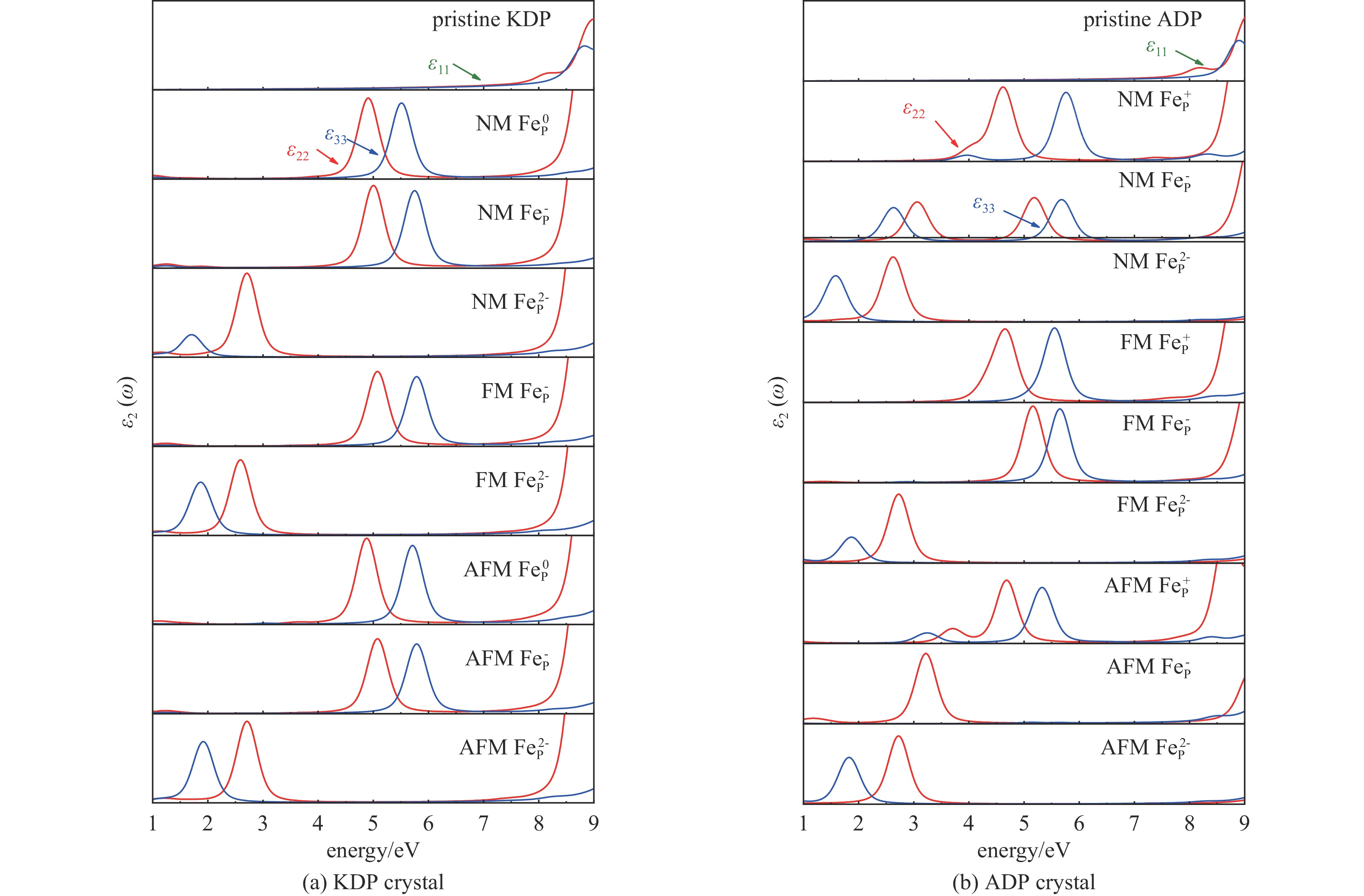
图 6 KDP和ADP晶体中不同磁性条件下FeP缺陷电荷态的介电函数虚部ε2 (ω)
Figure 6. Imaginary part of the dielectric function ε2 (ω) of charged FeP defects with different magnetic states in KDP and ADP crystals
表 1 KDP晶体中Fe取代不同位点的模型体系的缺陷形成能
Table 1. Defect formation energies of the KDP crystal with Fe in different locations
location defect formation energy/eV FeH 8.7 FeP 2.74 FeK 12.59 Fei 16.10  下载: 导出CSV
下载: 导出CSV
表 2 KDP和ADP晶体中Fe团簇缺陷的缺陷形成能
Table 2. Defect formation energies of the KDP crystal with Fe atom in different locations
defect defect formation energy/eV KDP ADP VOH 8.56 0.15 FeP+VOH 24.38 16.41
下载: 导出CSV
表 3 KDP和ADP晶体不同磁性条件下Fe−O键的键长变化
Table 3. Fe−O bond lengths in KDP and ADP with different magnetic states
defects bond length of Fe−O/nm KDP ADP pristine P-O 0.155 0.155 NM FeP+ 0.162 0.160 NM FeP0 0.163 0.162 NM FeP− 0.166 0.165 NM FeP2− 0.170 0.169 FM FeP+ 0.162 0.160 FM FeP0 0.163 0.162 FM FeP− 0.166 0.165 FM FeP2− 0.191 0.169 AFM FeP+ 0.162 0.161 AFM FeP0 0.163 0.162 AFM FeP− 0.166 0.165 AFM FeP2− 0.170 0.169
下载: 导出CSV
-
[1] Zaitseva N, Atherton J, Rozsa R, et al. Design and benefits of continuous filtration in rapid growth of large KDP and DKDP crystals[J]. Journal of Crystal Growth, 1999, 197(4): 911-920. doi: 10.1016/S0022-0248(98)01095-1 [2] Rashkovich L N, Kronsky N V. Influence of Fe3+ and Al3+ ions on the kinetics of steps on the {1 0 0} faces of KDP[J]. Journal of Crystal Growth, 1997, 182(3/4): 434-441. [3] Sasaki T, Yokotani A. Growth of large KDP crystals for laser fusion experiments[J]. Journal of Crystal Growth, 1990, 99(1/4): 820-826. [4] Eremina T A, Kuznetsov V A, Eremin N N, et al. On the mechanism of impurity influence on growth kinetics and surface morphology of KDP crystals—II: experimental study of influence of bivalent and trivalent impurity ions on growth kinetics and surface morphology of KDP crystals[J]. Journal of Crystal Growth, 2005, 273(3/4): 586-593. [5] Pritula I M, Velikhov Y N. Some aspects of UV absorption of NLO KDP crystals[C]//Proceedings of SPIE 3793, Operational Characteristics and Crystal Growth of Nonlinear Optical Materials. 1999: 202-208. [6] Salo V I, Kolybayeva M I, Puzikov V M, et al. Effect of impurities on the value of the bulk laser damage threshold of KDP single crystals[C]//Proceedings of SPIE 3359, Optical Diagnostics of Materials and Devices for Opto-, Micro-, and Quantum Electronics 1997. 1998: 549-552. [7] Salo V I, Atroschenko L V, Garnov S V, et al. Structure, impurity composition, and laser damage threshold of the subsurface layers in KDP and KD*P single crystals[C]//Proceedings of SPIE 2714, 27th Annual Boulder Damage Symposium: Laser-Induced Damage in Optical Materials. 1996: 197-201. [8] Azarov V V, Atroshchenko L V, Danileiko Y K, et al. Influence of structure defects on the internal optical strength of KDP single crystals[J]. Soviet Journal of Quantum Electronics, 1985, 15(1): 89-90. doi: 10.1070/QE1985v015n01ABEH005862 [9] Garces N Y, Stevens K T, Halliburton L E, et al. Optical absorption and electron paramagnetic resonance of Fe ions in KDP crystals[J]. Journal of Crystal Growth, 2001, 225(2/4): 435-439. [10] Eremina T A, Kuznetsov V A, Okhrimenko T M, et al. Structures of impurity defects in KDP and their influence on quality of crystals[C]//Proceedings of SPIE - The International Society for Optical Engineering. 2003: 153-158. [11] Malekfar R, Naghavi N. Raman back-scattering spectroscopy of KDP crystal doped by Fe+3 at low temperatures in the lower hydrogen bonds region[J]. AIP Conference Proceedings, 2007, 935(1): 126-131. [12] Endert G, Martin M L. The effect of chromium impurities on the laser damage threshold of KDP crystals[J]. Kristall und Technik, 1981, 16(5): K65-K66. doi: 10.1002/crat.19810160519 [13] 王波, 王圣来, 房昌水, 等. Fe3+对KDP晶体生长影响的研究[J]. 人工晶体学报, 2005, 34(2):205-208 doi: 10.3969/j.issn.1000-985X.2005.02.003Wang Bo, Wang Shenglai, Fang Changshui, et al. Effects of Fe3+ ion on the growth habit of KDP crystal[J]. Journal of Synthetic Crystals, 2005, 34(2): 205-208 doi: 10.3969/j.issn.1000-985X.2005.02.003 [14] 郭德成. KDP晶体微观缺陷检测及辐照效应研究[D]. 成都: 电子科技大学, 2016Guo Decheng. Radiation effects and micro defects detection of KDP crystals[D]. Chengdu: University of Electronic Science and Technology of China, 2016 [15] Liu Yongqiang, Li Xiangcao, Wu Jian, et al. First-principles studies on the electronic and optical properties of Fe-doped potassium dihydrogen phosphate crystal[J]. Computational Materials Science, 2018, 143: 398-402. doi: 10.1016/j.commatsci.2017.11.035 [16] Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set[J]. Computational Materials Science, 1996, 6(1): 15-50. doi: 10.1016/0927-0256(96)00008-0 [17] Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Physical Review B, 1996, 54(16): 11169-11186. doi: 10.1103/PhysRevB.54.11169 [18] Monkhorst H J, Pack J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13(12): 5188-5192. doi: 10.1103/PhysRevB.13.5188 [19] Liu C S, Kioussis N, Demos S G, et al. Electron- or hole-assisted reactions of H defects in hydrogen-bonded KDP[J]. Physical Review Letters, 2003, 91: 015505. doi: 10.1103/PhysRevLett.91.015505 [20] Liu C S, Hou C J, Kioussis N, et al. Electronic structure calculations of an oxygen vacancy in KH2PO4[J]. Physical Review B, 2005, 72: 134110. doi: 10.1103/PhysRevB.72.134110 [21] Liu C S, Zhang Q, Kioussis N, et al. Electronic structure calculations of intrinsic and extrinsic hydrogen point defects in KH2PO4[J]. Physical Review B, 2003, 68: 224107. doi: 10.1103/PhysRevB.68.224107 [22] Wang Kunpeng, Fang Changshui, Zhang Jianxiu, et al. First-principles study of interstitial oxygen in potassium dihydrogen phosphate crystals[J]. Physical Review B, 2005, 72: 184105. doi: 10.1103/PhysRevB.72.184105 [23] Nikogosyan D N. Nonlinear optical crystals: a complete survey[M]. New York: Springer, 2005: 119. [24] Dhanaraj P V, Bhagavannarayana G, Rajesh N P. Effect of amino acid additives on crystal growth parameters and properties of ammonium dihydrogen orthophosphate crystals[J]. Materials Chemistry and Physics, 2008, 112(2): 490-495. doi: 10.1016/j.matchemphys.2008.06.003 [25] Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865-3868. doi: 10.1103/PhysRevLett.77.3865 [26] Heyd J, Scuseria G E, Ernzerhof M. Hybrid functionals based on a screened Coulomb potential[J]. The Journal of Chemical Physics, 2003, 118(18): 8207-8215. doi: 10.1063/1.1564060 [27] Krukau A V, Vydrov O A, Izmaylov A F, et al. Influence of the exchange screening parameter on the performance of screened hybrid functionals[J]. The Journal of Chemical Physics, 2006, 125: 224106. doi: 10.1063/1.2404663 [28] Silvestrelli P L. Van der Waals interactions in DFT made easy by Wannier functions[J]. Physical Review Letters, 2008, 100: 053002. doi: 10.1103/PhysRevLett.100.053002 [29] Silvestrelli P L. Van der Waals interactions in density functional theory using Wannier functions[J]. The Journal of Physical Chemistry A, 2009, 113(17): 5224-5234. doi: 10.1021/jp811138n [30] Andrinopoulos L, Hine N D M, Mostofi A A. Calculating dispersion interactions using maximally localized Wannier functions[J]. The Journal of Chemical Physics, 2011, 135: 154105. doi: 10.1063/1.3647912 [31] 张克从, 王希敏. 非线性光学晶体材料科学[M]. 2版. 北京: 科学出版社, 2005Zhang Kecong, Wang Ximing. Nonlinear optical crystal materials[M]. 2nd ed. Beijing: Science Press, 2005 [32] Sui Tingting, Wan Chubin, Xu Mingxia, et al. Hybrid density functional theory for the stability and electronic properties of Fe-doped cluster defects in KDP crystal[J]. CrystEngComm, 2021, 23(44): 7839-7845. doi: 10.1039/D1CE01140E [33] 杨华. 3d过渡金属氧化物电子结构的第一性原理计算[D]. 天津: 天津大学, 2013Yang Hua. First principles calculations on electronic structure of 3d transition metal oxides[D]. Tianjin: Tianjin University, 2013 [34] 王娟. 基于第一性原理计算的半金属磁性材料电子结构和自旋结构研究[D]. 武汉: 武汉理工大学, 2007Wang Juan. First-principle study on the electronic structure and spin structure of half-metallic ferromagnets[D]. Wuhan: Wuhan University of Technology, 2007 [35] Zheng Haiwu, Zhang Yongjia, Yan Yuli, et al. Experimental observation and theoretical calculation of magnetic properties in Fe-doped cubic SiC nanowires[J]. Carbon, 2014, 78: 288-297. doi: 10.1016/j.carbon.2014.07.005 [36] Duan Yuping, Liu Zhuo, Zhang Yahong, et al. A theoretical study of the dielectric and magnetic responses of Fe-doped α-MnO2 based on quantum mechanical calculations[J]. Journal of Materials Chemistry C, 2013, 1(10): 1990-1994. doi: 10.1039/c3tc00902e [37] Garces N Y, Stevens K T, Halliburton L E, et al. Identification of electron and hole traps in KH2PO4 crystals[J]. Journal of Applied Physics, 2001, 89(1): 47-52. doi: 10.1063/1.1320030 [38] Sui Tingting, Lian Yafei, Xu Mingxia, et al. Stability and electronic structure of hydrogen vacancies in ADP: hybrid DFT with vdW correction[J]. RSC Advances, 2018, 8(13): 6931-6939. doi: 10.1039/C7RA13212C -




 下载:
下载:







 点击查看大图
点击查看大图
计量
- 文章访问数: 810
- HTML全文浏览量: 302
- PDF下载量: 71
- 被引次数: 0
