

Adsorption behavior of water molecules on the surface of lithium hydride
-
摘要: 运用理论分析方法计算研究了水分子在氢化锂表面的吸附行为,分析了氢化锂表面改性对其疏水性能的影响。结果表明,在LiH-111面和LiH-100面上构建槽结构、柱状结构后,水分子在其上的吸附力比完整表面更强,说明表面微结构的引入的确改变了势能分布。壁相交处存在势能叠加,加强了吸附水分子的能力,但是没有引起表面的亲水性能变化。水分子可以稳定的吸附在完美的LiH(001)表面,其解离能垒仅为0.386 eV,这一解离反应在室温下完全可以进行。水分子极易在具有结构缺陷的LiH表面解离,这是LiH在一定湿度的空气和水环境中极易分解的根本原因。Abstract: The theoretical analysis method is used to calculate the adsorption behavior of water molecules on the surface of lithium hydride, and analyze the influence of surface modification of lithium hydride on its hydrophobic performance. The results show that after constructing groove structure and columnar structure on LiH-111 surface and LiH-100 surface, the adsorption force to water molecules of the modified surface is stronger than that of the complete surface, indicating that the introduction of surface microstructure does change the potential energy distribution. There is a superposition of potential energy at the intersection of the walls, which strengthens the ability to adsorb water molecules, but does not cause changes in the hydrophilic properties of the surface. Water molecules can be stably adsorbed on the perfect LiH (001) surface, and its dissociation energy barrier is only 0.386 eV. This dissociation reaction can be carried out at room temperature. Water molecules are easily dissociated on the LiH surface with structural defects, which is the fundamental reason why LiH decomposes easily in a certain humidity air and water environment.
-
Key words:
- lithium hydride /
- adsorption behavior /
- hydrophobicity /
- kinetics
-
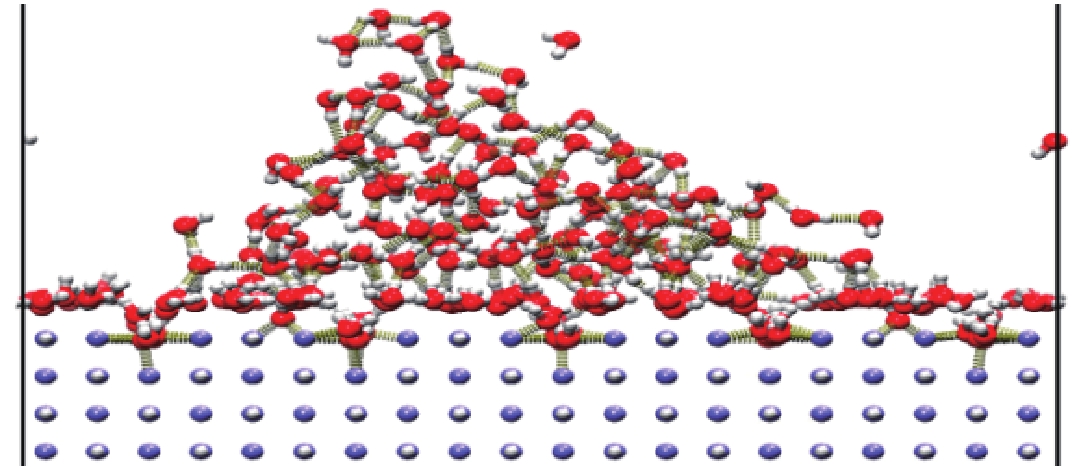
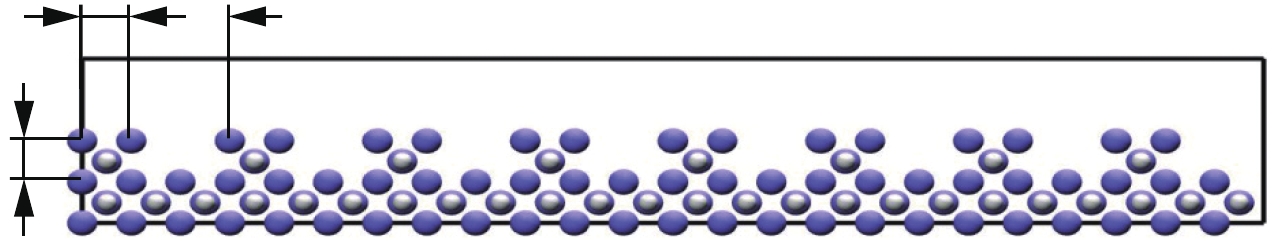
图 7 LiH-100面槽结构与水的相互作用
Figure 7. Interaction between LiH-100 surface grooves and water
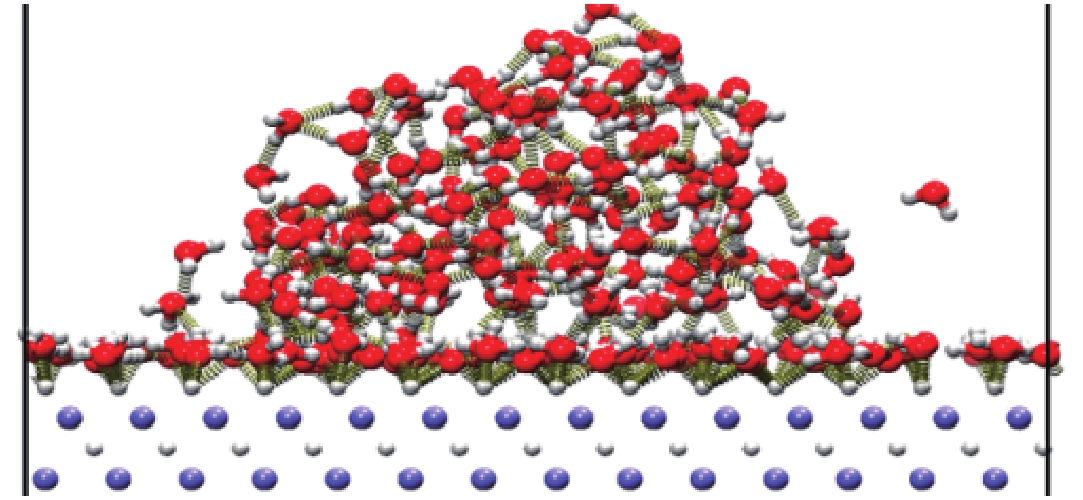
图 8 LiH-100面柱结构与水的相互作用
Figure 8. Interaction between LiH-100 surface columns and water

图 9 LiH-111面槽结构与水的相互作用
Figure 9. Interaction between LiH-111 surface grooves and water
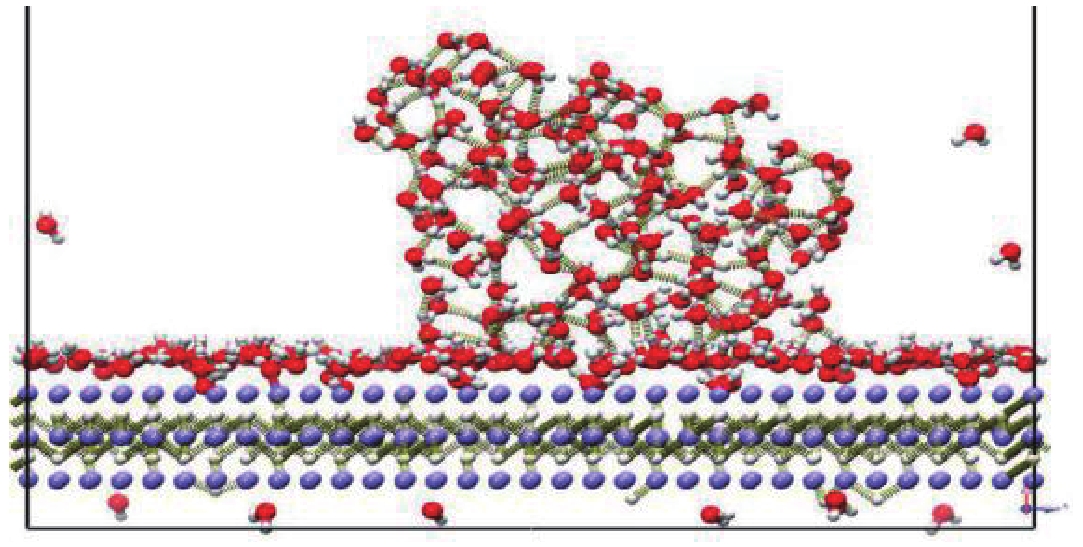
图 10 LiH-111面柱结构与水的相互作用
Figure 10. Interaction between LiH-111 surface columns and water
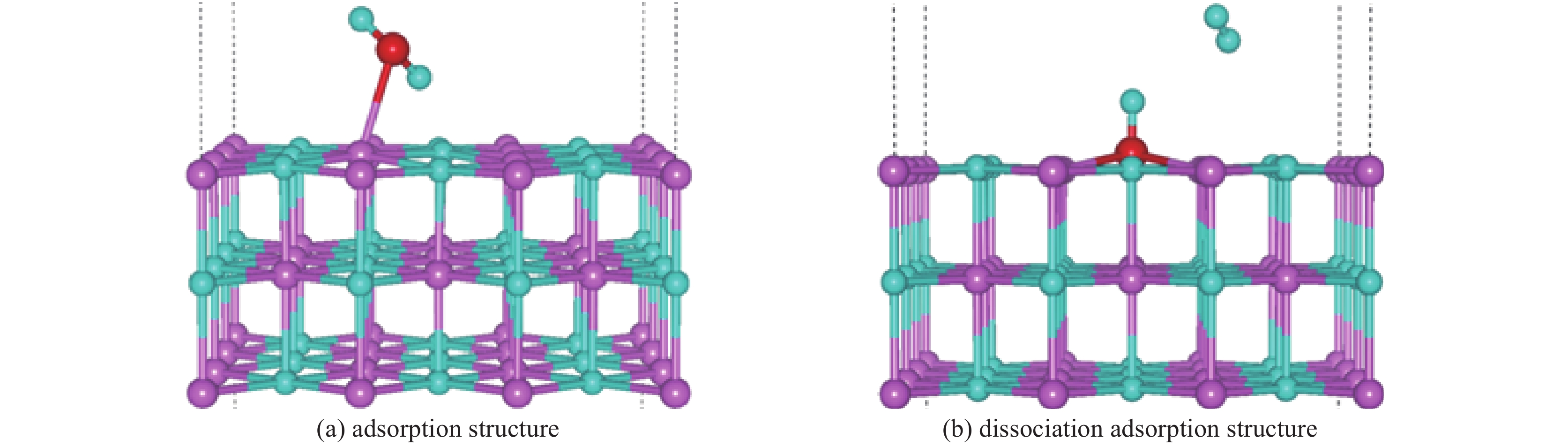
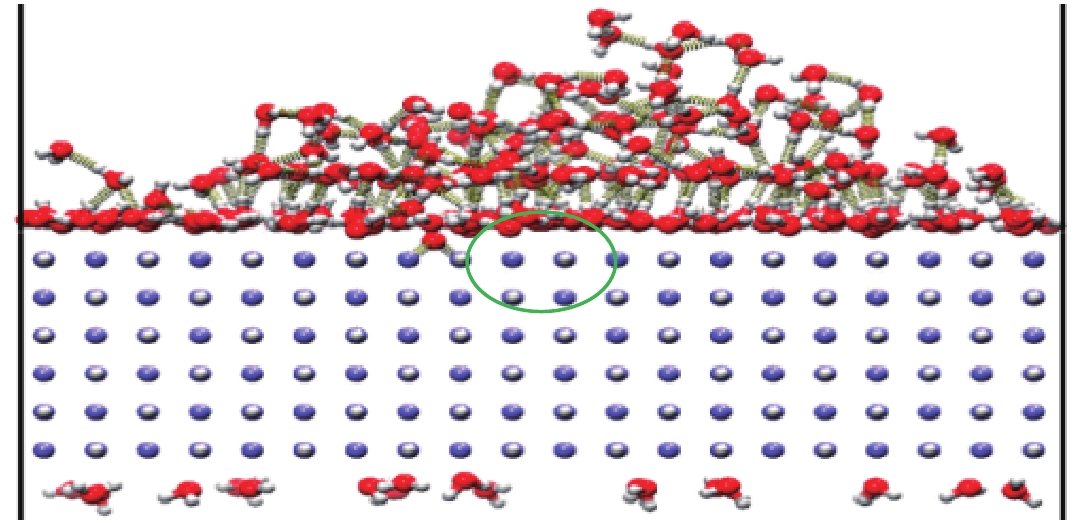
图 12 水分子在完美的LiH(001)表面的吸附行为
Figure 12. Adsorption behavior of water molecules on a perfect LiH (001) surface
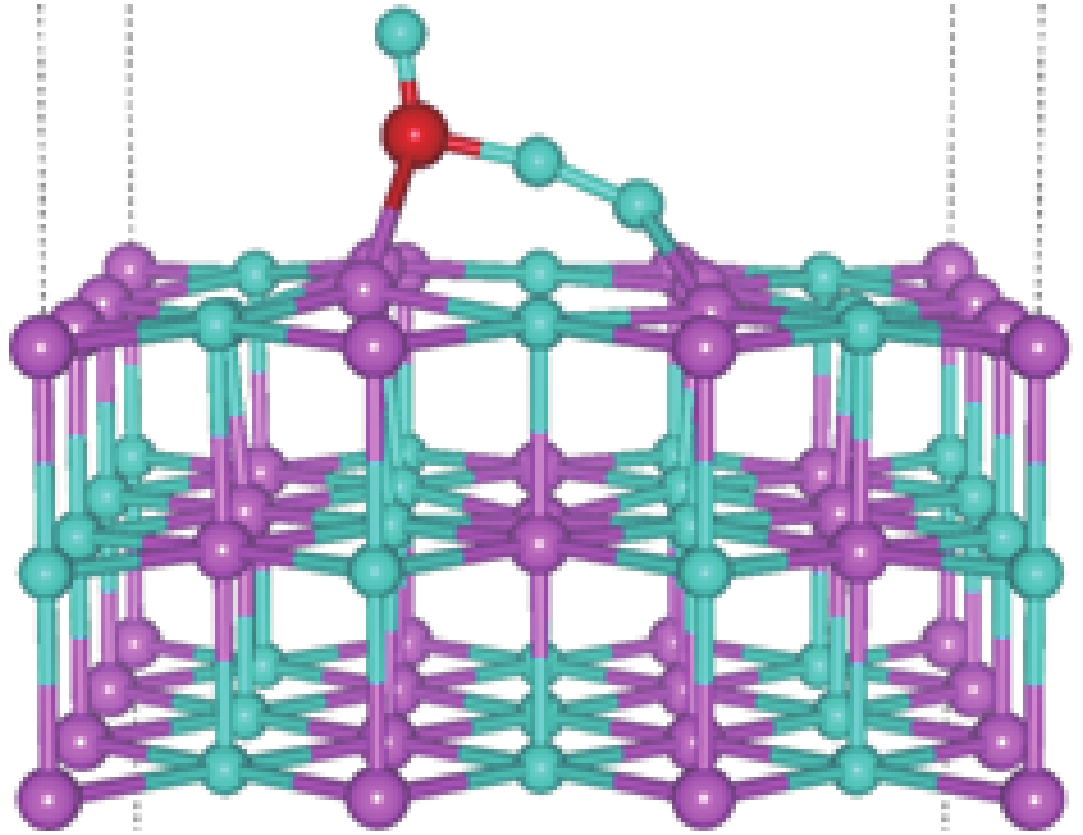
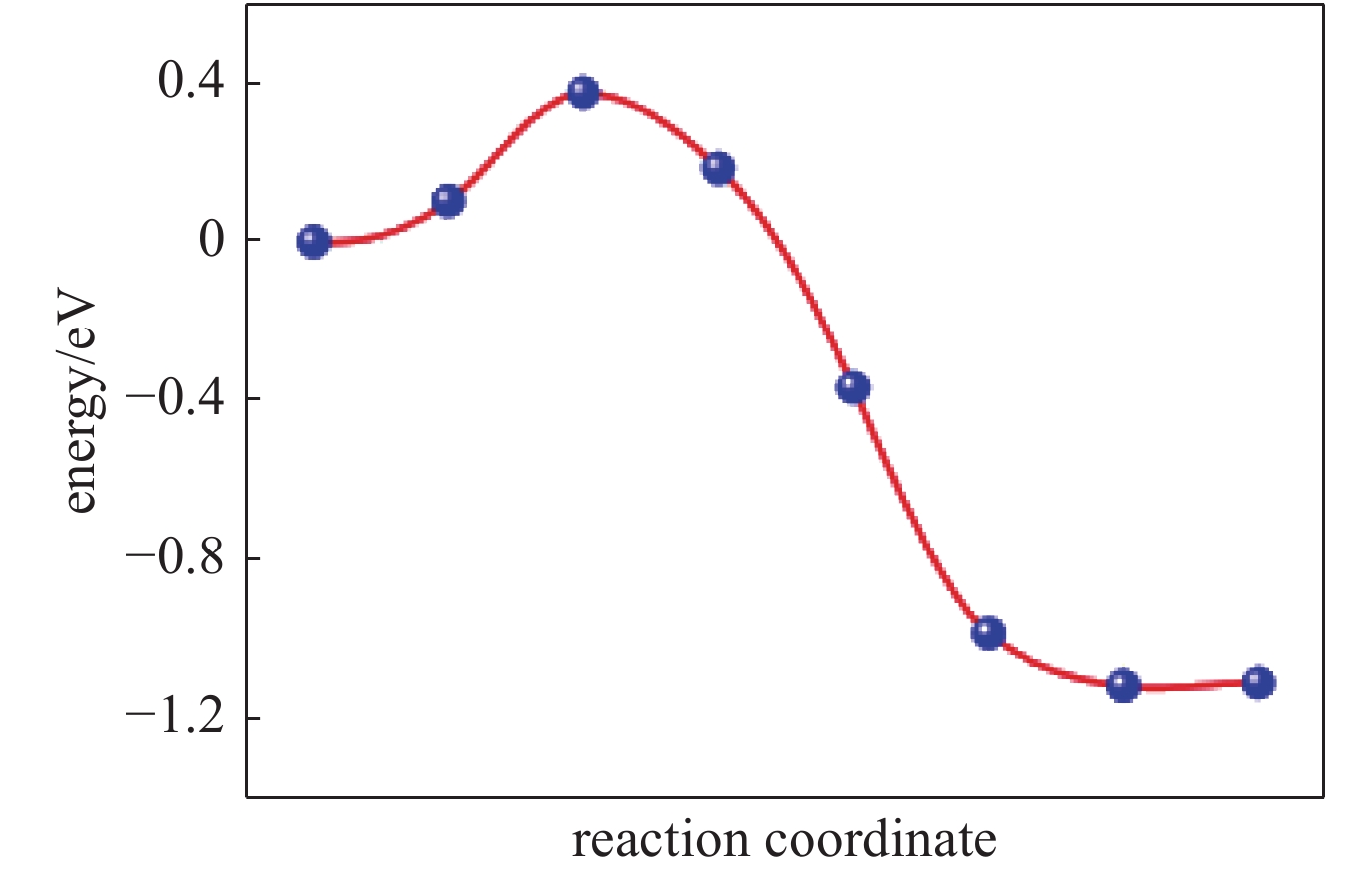
图 13 水分子在完美的LiH(001)表面解离反应的过渡态结构
Figure 13. Transition state structure of the dissociation reaction of water molecules on the perfect LiH (001) surface
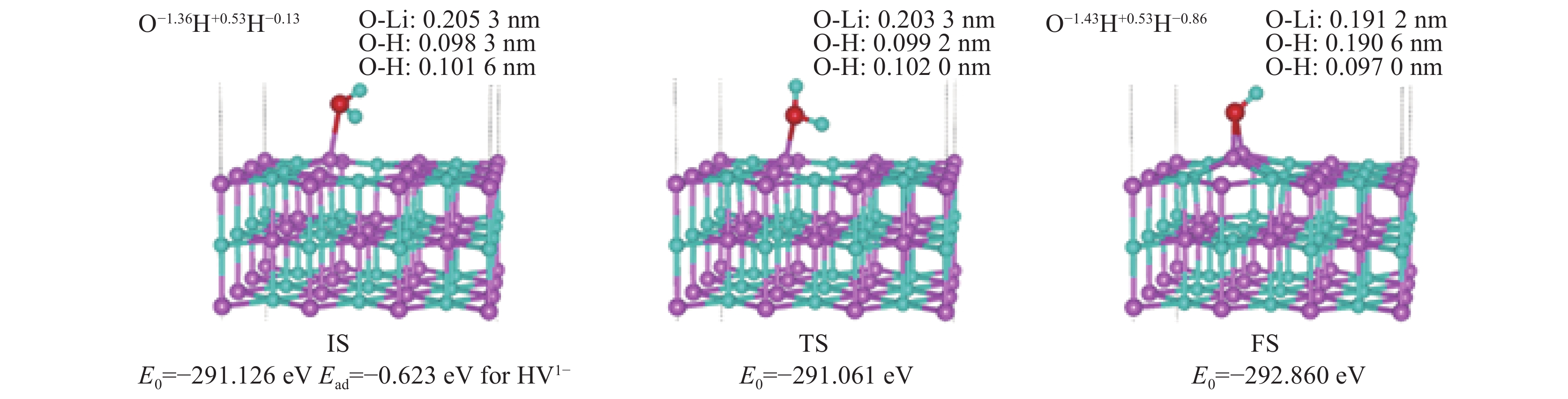
图 15 水分子在带有一个负电荷的氢空位的LiH(001)表面的吸附行为
Figure 15. Adsorption behavior of water molecules on LiH (001) surface with a negatively charged hydrogen vacancy
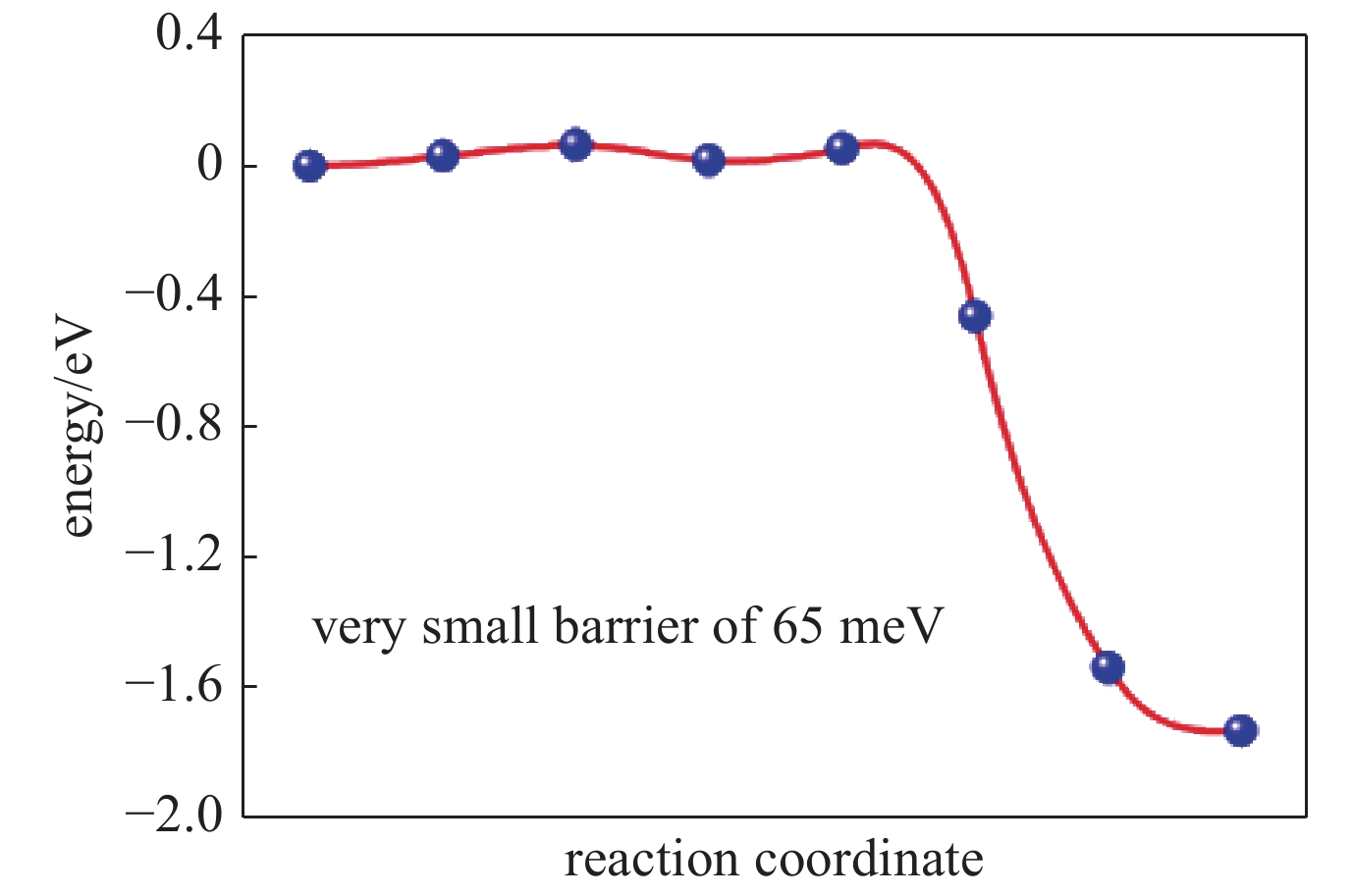
图 16 水分子在带有一个负电荷的氢空位的LiH(001)表面的解离路径
Figure 16. Dissociation path of water molecules on LiH (001) surface with a negatively charged hydrogen vacancy
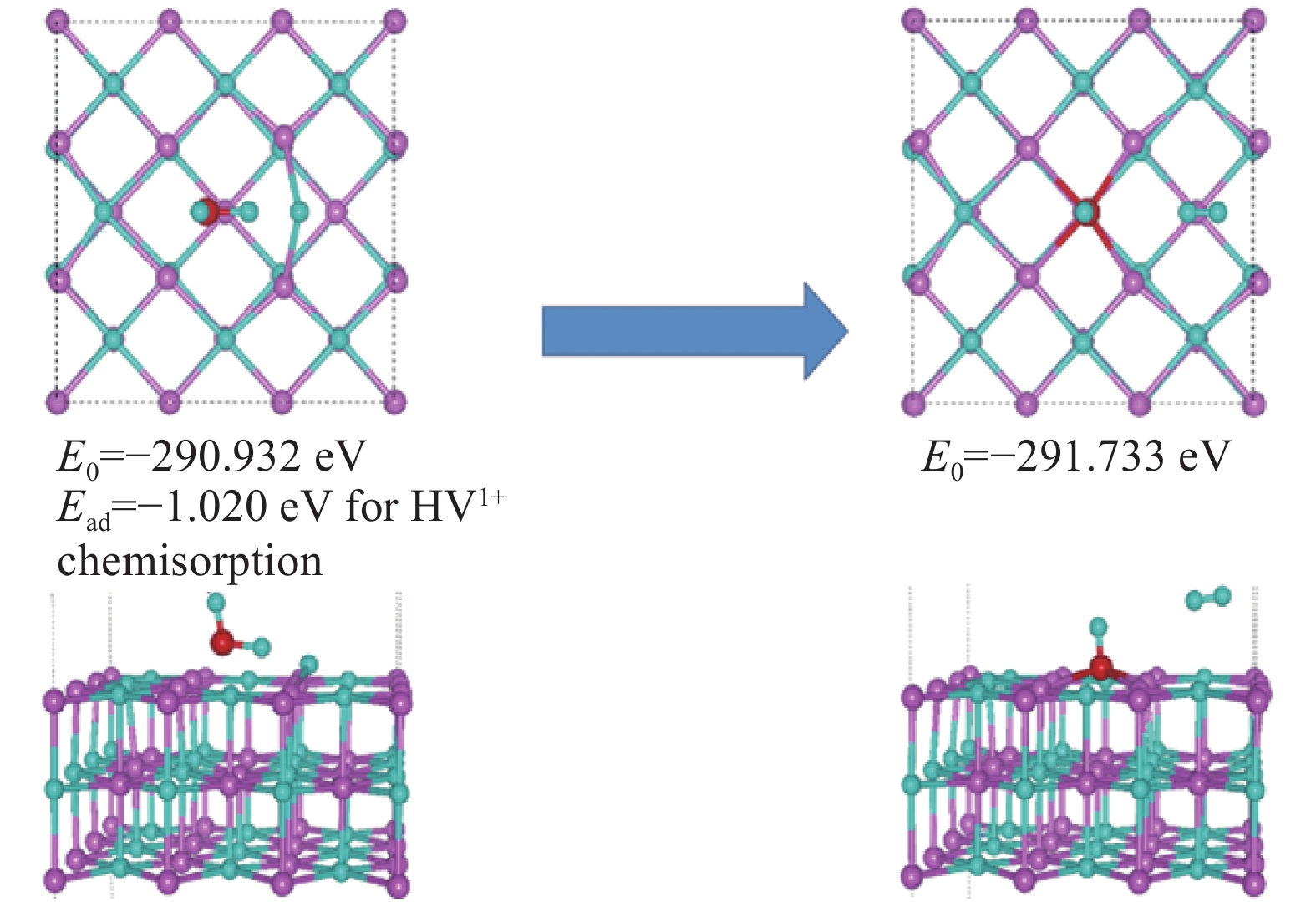
图 17 水分子在带有一个正电荷的氢空位的LiH(001)表面的吸附行为
Figure 17. Adsorption behavior of water molecules on LiH (001) surface with a positively charged hydrogen vacancy
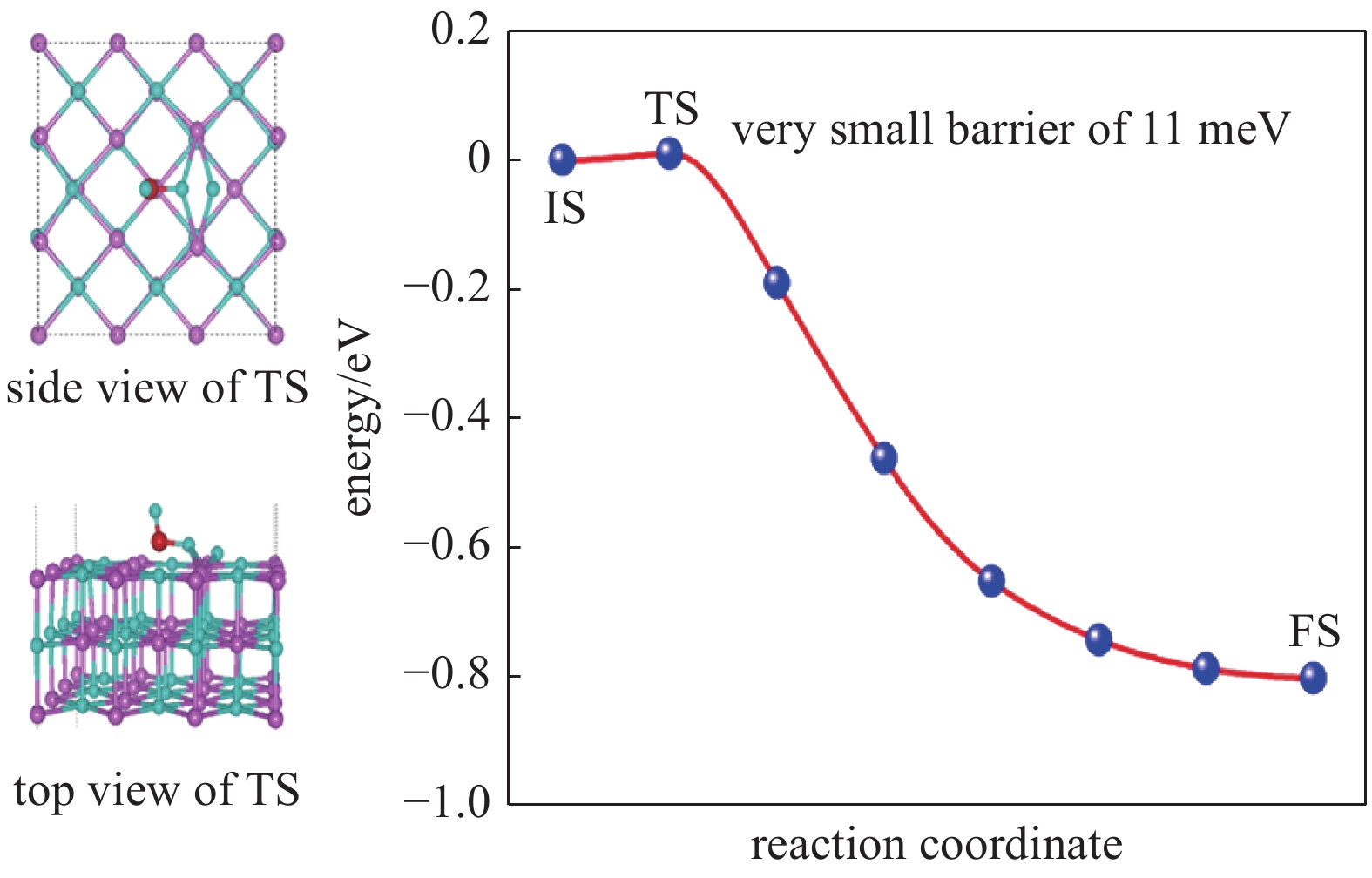
图 18 水分子在带有一个正电荷的氢空位的LiH(001)表面的解离路径
Figure 18. Dissociation path of water molecules on LiH (001) surface with a positively charged hydrogen vacancy
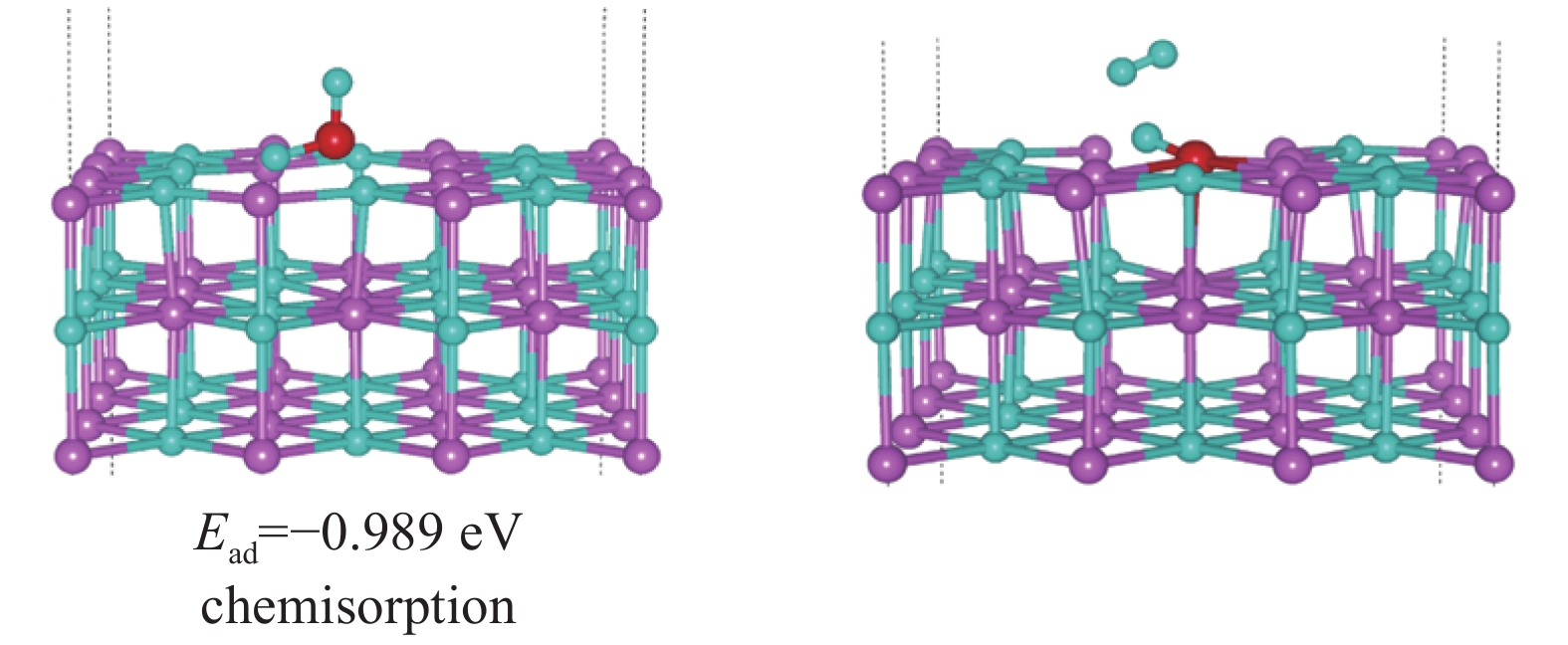
图 19 水分子在表面存在Li-H双空位时的LiH(001)表面的吸附行为
Figure 19. Adsorption behavior of LiH (001) surface when water molecules have Li-H double vacancies on the surface
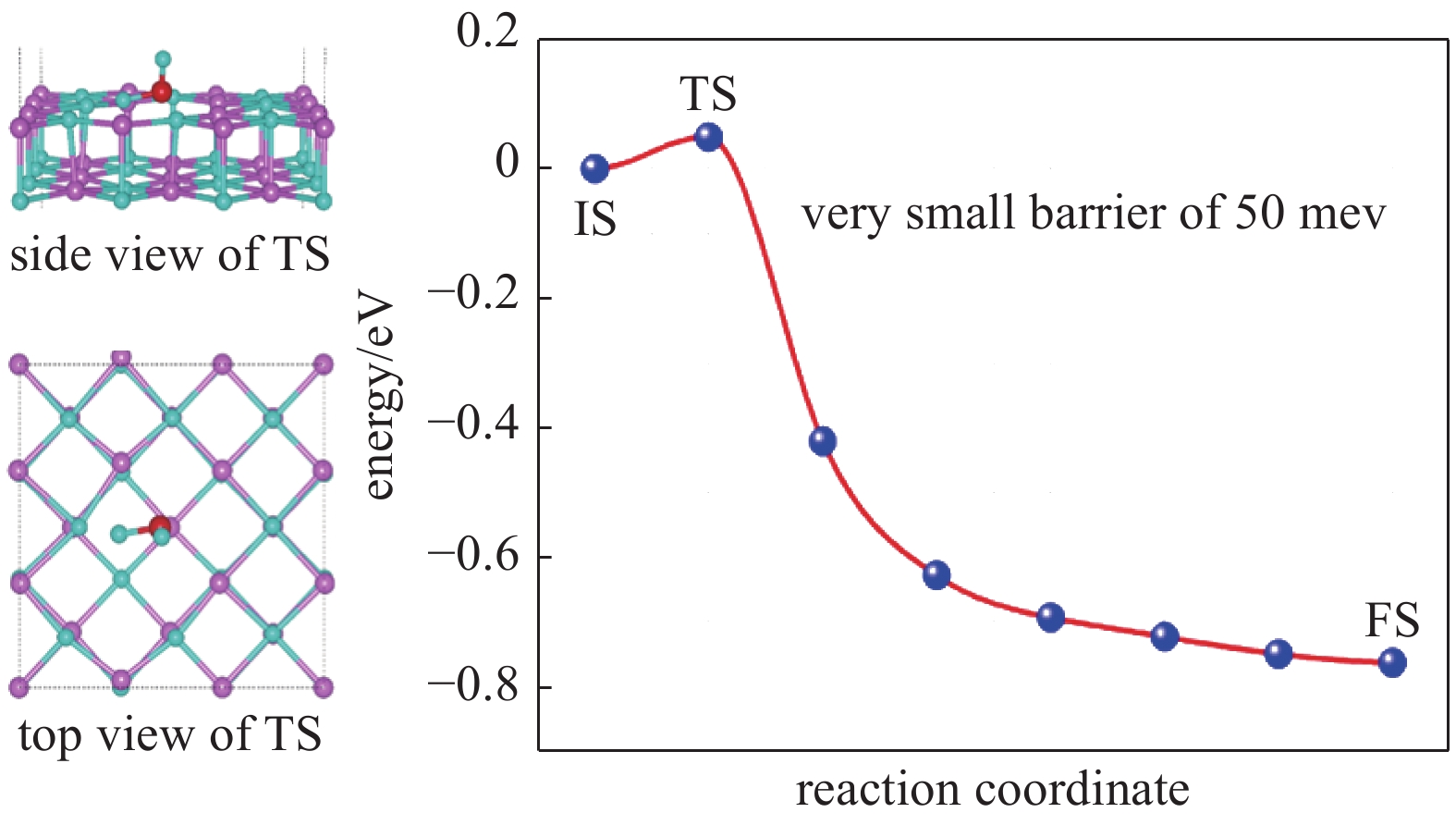
图 20 水分子在表面存在Li-H双空位时的LiH(001)表面的解离路径
Figure 20. Dissociation path of LiH (001) surface when water molecules have Li-H double vacancies on the surface
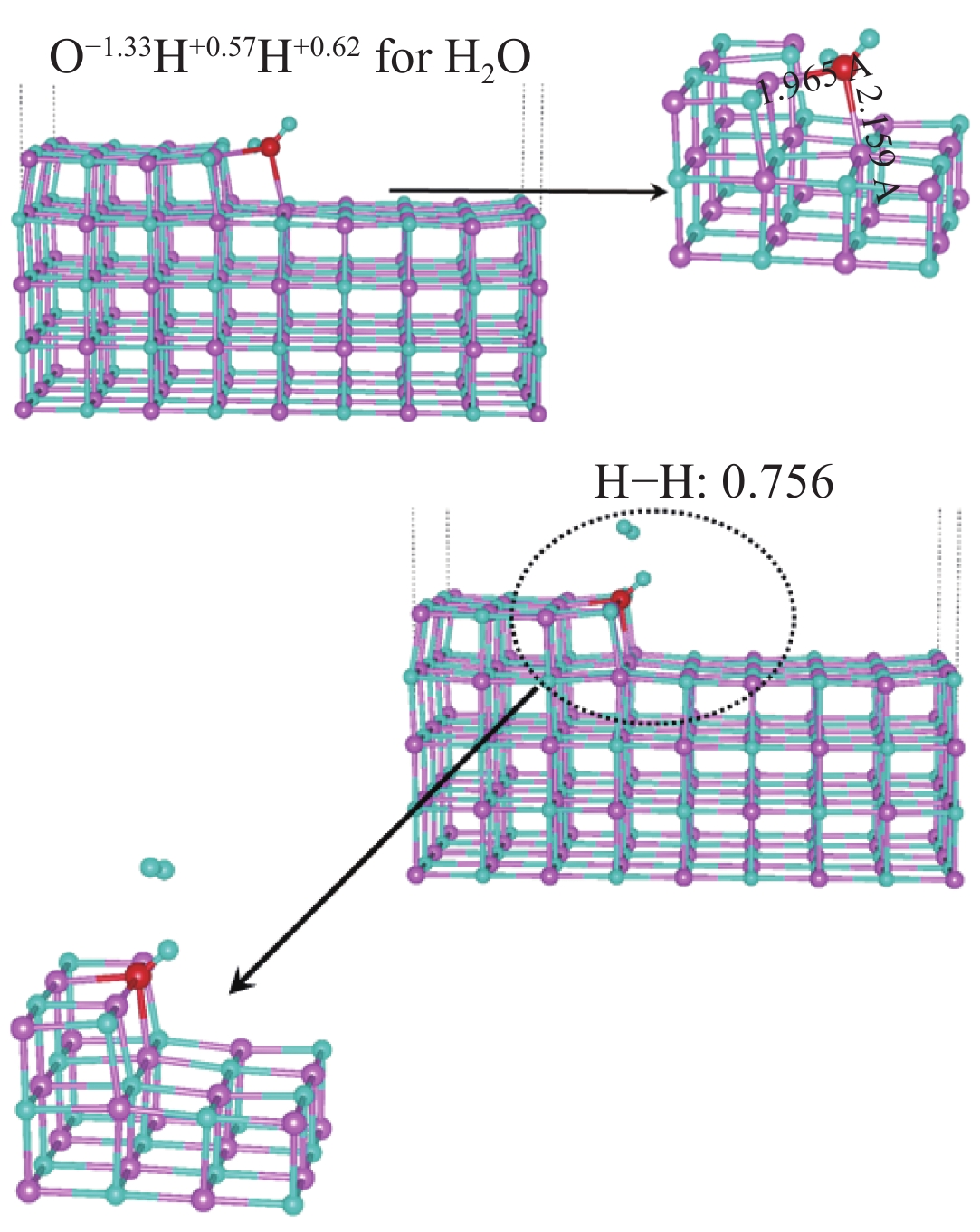
图 21 水分子在LiH(001)表面台阶处的吸附过程
Figure 21. Adsorption process of water molecules on the LiH (001) surface steps
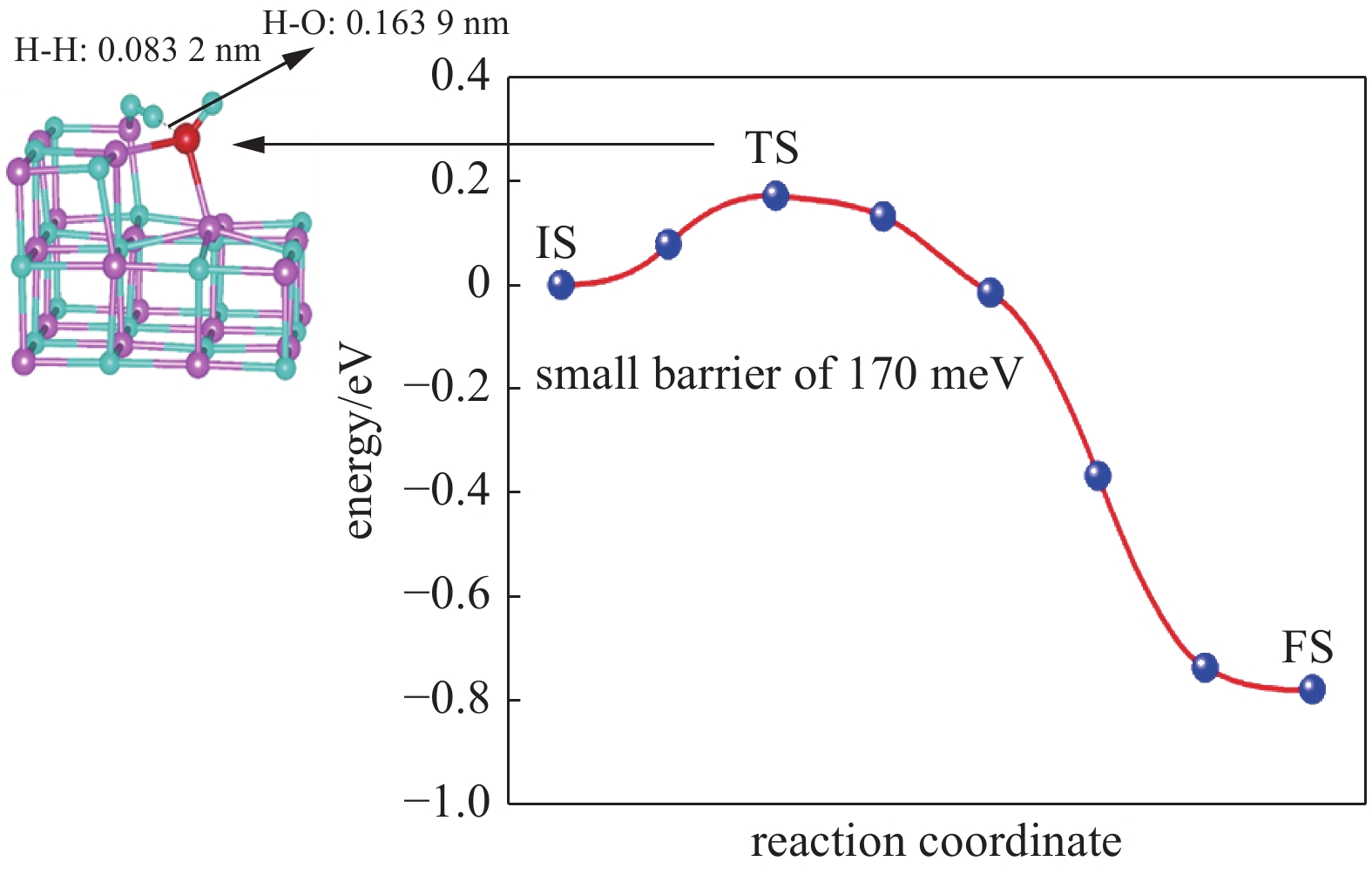
图 22 水分子在LiH(001)表面台阶处的解离路径
Figure 22. Dissociation path of water molecules at the steps of LiH (001) surface
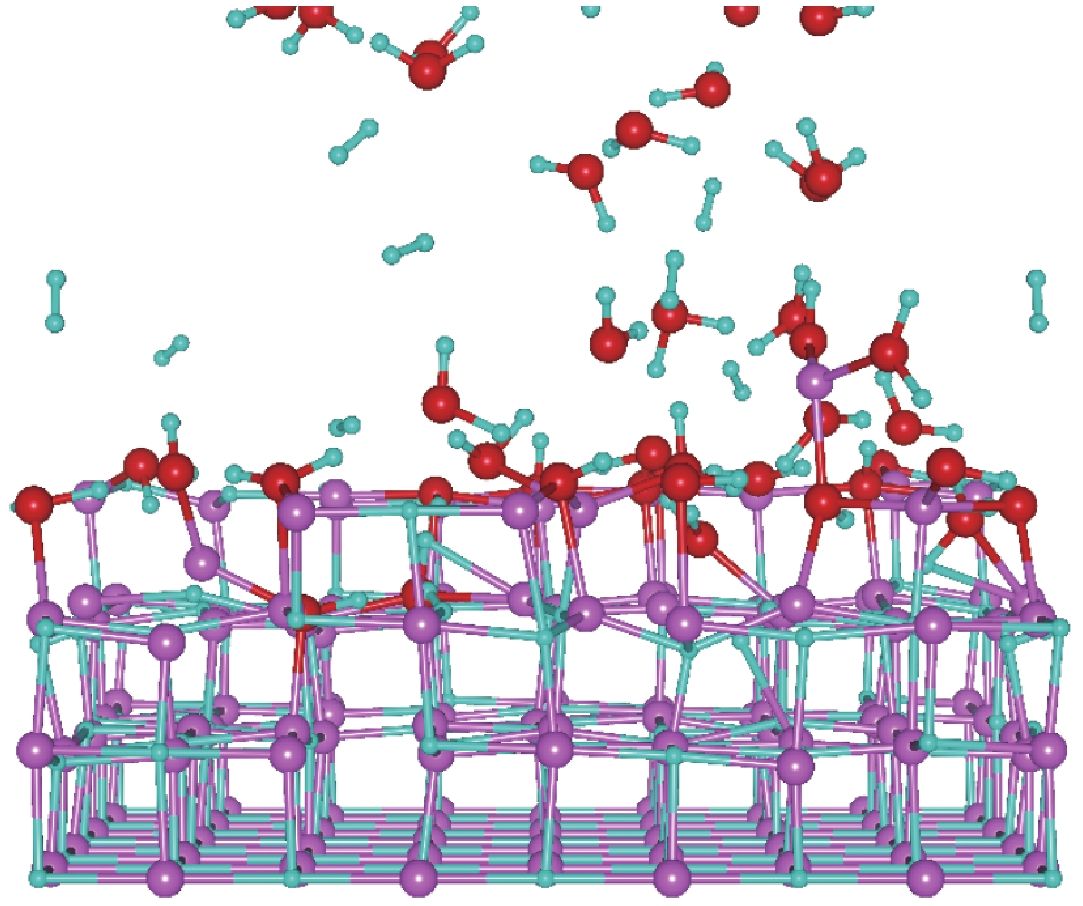
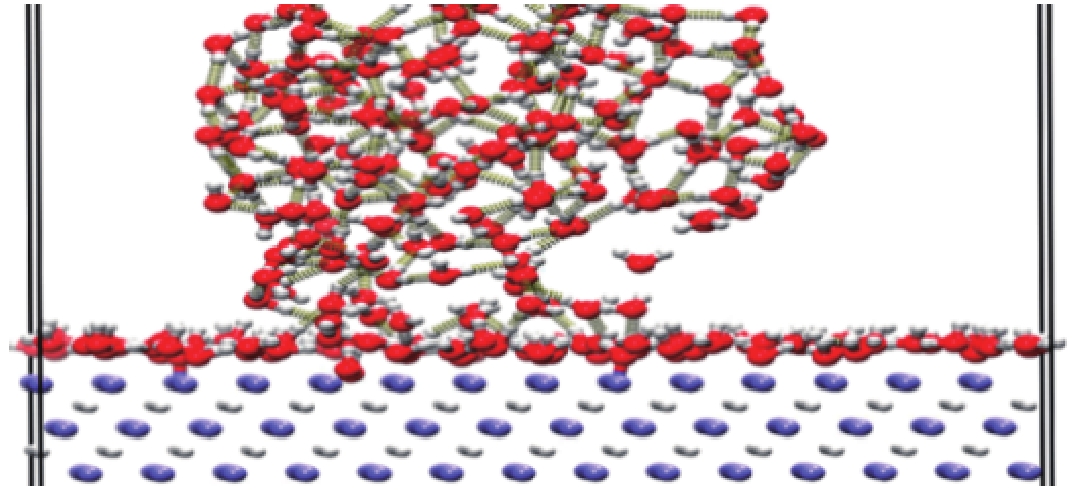
图 23 水分子在存在缺陷的LiH(001)表面的吸附过程
Figure 23. Adsorption process of water molecules on a defective LiH (001) surface
-
[1] Haertling C, Hanrahan R J, Smith R. A literature review of reactions and kinetics of lithium hydride hydrolysis[J]. Journal of Nuclear Materials, 2006, 349(1/2): 195-233. [2] Chen P, Xiong Z, Luo J, et al. Interaction between lithium amide and lithium hydride[J]. Journal of Physical Chemistry B, 2003, 107(39): 10967-10970. doi: 10.1021/jp034149j [3] Kojima Y, Kawai Y, Kimbara M, et al. Hydrogen generation by hydrolysis reaction of lithium borohydride[J]. International Journal of Hydrogen Energy, 2004, 29(12): 1213-1217. doi: 10.1016/j.ijhydene.2003.12.009 [4] Stober, K. J, Cantwell B J, Otaibi R A. Hypergolic ignition of lithium–aluminum–hydride-doped paraffin wax and nitric acid[J]. Journal of Propulsion and Power, 2020, 36(3): 435-445. doi: 10.2514/1.B37425 [5] Houk K N, Rondan N G, Schleyer P V, et al. Transition structures for additions of lithium hydride and methyllithium to ethylene and acetylene[J]. Journal of the American Chemical Society, 1985, 107(9): 2821-2823. doi: 10.1021/ja00295a053 [6] Kang X, Fang Z, Kong L, et al. Ammonia borane destabilized by lithium hydride: an advanced on-board hydrogen storage material[J]. Advanced Materials, 2008, 20(14): 2756-2759. doi: 10.1002/adma.200702958 [7] Weber G, Sciora E, Guichard J, et al. New insight on the lithium hydride–water vapor reaction system[J]. International Journal of Hydrogen Energy, 2018, 43(50): 22557-22567. doi: 10.1016/j.ijhydene.2018.10.089 [8] Dovesi R, Ermondi C, Ferrero E, et al. Hartree-Fock study of lithium hydride with the use of a polarizable basis set[J]. Physical Review B, 1984, 29(6): 3591-3600. doi: 10.1103/PhysRevB.29.3591 [9] 胡玉坤, 丁静, 彭晓峰, 等. 锂离子交换ZSM-5型分子筛中水分子吸附特性的分子模拟[J]. 硅酸盐学报, 2007(9):1247-1252. (Hu Yukun, Ding Jing, Peng Xiaofeng, et al. Molecular simultion of characteristics for water adsorption on ZSM-5 type zeolite doped by lithium ion[J]. Journal of the Chinese Ceramic Society, 2007(9): 1247-1252 doi: 10.3321/j.issn:0454-5648.2007.09.022 [10] 吕岩, 王涛, 马卫华. 基于密度泛函理论研究水在磷酸锂(100)表面的吸附[J]. 广州化工, 2016, 44(9):1-4. (Lü Yan, Wang Tao, Ma Weihua. Adsorption of water on Li3PO4 (100) surface from density functional theory[J]. Guangzhou Chemical Industry, 2016, 44(9): 1-4 doi: 10.3969/j.issn.1001-9677.2016.09.001 [11] 王林林, 朱灵燕, 刘跃龙, 等. 混合捕收剂在锂云母表面吸附行为的分子动力学模拟研究[J]. 有色金属(选矿部分), 2019(2):108-114. (Wang Linlin, Zhu Lingyan, Liu Yuelong, et al. Molecular dynamics simulation study on adsorption behavior of mixed collector on lithium mica surface[J]. Nonferrous Metals Mineral Processing Section, 2019(2): 108-114 [12] 李璐, 李奕, 郭欣, 等. 水在HfO2(111)和(110)表面的吸附与解离[J]. 物理化学学报, 2013(5):61-69. (Li Lu, Li Yi, Guo Xin, et al. Adsorption and dissociation of water on HfO2(111) and (110) surfaces[J]. Acta Physico-Chimica Sinica, 2013(5): 61-69 [13] 刘够生, 宋兴福, 汪瑾, 等. 水分子在MoO3原子簇模型表面吸附的密度泛函研究[J]. 分子催化, 2005, 19(2):136-140. (Liu Gousheng, Song Xingfu, Wang Jin, et al. Density functional theoretical study of water molecular adsorption on the surface of MoO3 with the cluster model[J]. Journal of Molecular Catalysis, 2005, 19(2): 136-140 doi: 10.3969/j.issn.1001-3555.2005.02.012 [14] 杜佳, 闵凡飞, 张明旭, 等. 水分子在铵伊利石表面吸附的密度泛函研究[J]. 中国矿业大学学报, 2017, 46(6):1349-1356. (Du Jia, Min Fanfei, Zhang Mingxu, et al. Mechanism of H2O adsorption on ammonium-illite surface based on density functional theory[J]. Journal of China University of Mining & Technology, 2017, 46(6): 1349-1356 [15] 薛严冰, 唐祯安, 孙伟民. 水分子在SnO2(110)表面吸附特性的密度泛函计算[J]. 大连交通大学学报, 2012, 33(5):93-96. (Xue Yanbing, Tang Zhen’an, Sun Weimin. Density functional calculation of properties of water molecule adsorption on SnO2 (110) surface[J]. Journal of Dalian Jiaotong University, 2012, 33(5): 93-96 doi: 10.3969/j.issn.1673-9590.2012.05.022 [16] 张瑶, 李照兵, 张鑫, 等. 水分子在Bi(111)表面上的吸附和自组装[J]. 中国科学(化学), 2016, 46(4):389-393. (Zhang Yao, Li Zhaobing, Zhang Xin, et al. Adsorption and self-assembly of water molecules on Bi (111)[J]. Scientia Sinica(Chimica), 2016, 46(4): 389-393 [17] 付纯鹤, 卢辉丽, 孙少瑞. 密度泛函理论方法研究6H-SiC(0001)表面对氧分子和水分子的吸附[J]. 化学物理学报, 2019(4):451-456. (Fu Chunhe, Lu Huili, Sun Shaorui. Density functional theory study for adsorption of oxygen and water molecules on 6H-SiC (0001) surface[J]. Chinese Journal of Chemical Physics, 2019(4): 451-456 [18] 赵红华, 江舒棋, 葛源源, 等. 不同阳离子基蒙脱石吸附水分子的分子动力学模拟分析[J]. 中国科学:技术科学, 2019, 49(6):703-715. (Zhao Honghua, Jiang Shuqi, Ge Yuanyuan, et al. Molecular dynamics simulation of water molecules adsorption by different cations based montmorillonite[J]. Scientia Sinica(Technologica), 2019, 49(6): 703-715 [19] Oshikiri M, Boero M. Water molecule adsorption properties on the BiVO4 (100) surface[J]. Journal of Physical Chemistry B, 2006, 110(18): 9188-9194. doi: 10.1021/jp0555100 [20] Kohtani M, Breaux G A, Jarrold M F, et al. Water molecule adsorption on protonated dipeptides[J]. Journal of the American Chemical Society, 2004, 126(4): 1206-1213. doi: 10.1021/ja0359557 [21] Rangel E, Ruizchavarria G, Magana L F, et al. Water molecule adsorption on a titanium–graphene system with high metal coverage[J]. Carbon, 2009, 47(2): 531-533. doi: 10.1016/j.carbon.2008.11.037 [22] Pang Z Q, Zhang Y, Rong Z, et al. Adsorption and dissociation of water on oxygen pre-covered Cu (110) observed with scanning tunneling microscopy[J]. Acta Physica Sinica, 2016, 65: 226801. [23] Wungu T D, Agusta M K, Saputro A G, et al. First principles calculation on the adsorption of water on lithium-montmorillonite (Li-MMT)[J]. Phys Condens Matter, 2012, 24: 475506. doi: 10.1088/0953-8984/24/47/475506 [24] Wang C, Zhang K, Song P, et al. First-principles study of nitrogen adsorption and dissociation on PuH2 (111) surface[J]. Molecules, 2020, 25(8): 1891-1903. doi: 10.3390/molecules25081891 -
 下载:
下载:



 点击查看大图
点击查看大图
计量
- 文章访问数: 2290
- HTML全文浏览量: 575
- PDF下载量: 69
- 被引次数: 0
